


 LongeCity
LongeCityAdvocacy & Research for Unlimited Lifespans
 Last Updated:
11 July 2026 - 11:40 PM
Last Updated:
11 July 2026 - 11:40 PM
Age-related impairment of drainage of cerebrospinal fluid from the brain is a topic of increasing interest. This was pioneered by the work of Leucadia Therapeutics, specifically focused on the drainage path for the olfactory bulb leading through the cribriform plate, but most present work is focused instead on the glymphatic system drainage that parallels the vasculature supplying the brain. The two pathways decline in capacity with age, but for very different reasons, and will need different forms of therapy. Thin channels for fluid flow through the cribriform plate ossify shut with age. The glymphatic system suffers from a failure of regulation of peristaltic flow through vessels, however.
Cerebrospinal fluid drainage is a way to remove metabolic waste from the brain. Declining flow means that this waste will build up. This includes the protein aggregates associated with neurodegenerative conditions, and probably a great many other forms of metabolic waste that individually have more subtle effects, but collectively act to provoke cell dysfunction when present at high levels. One of the more important consequences is thought to be a maladaptive inflammatory reaction in glial cells in the brain, which in turn drives the onset and progression of neurodegenerative conditions.
In today's open access paper, researchers assess measures indicative of loss of cerebrospinal fluid drainage in patients with isolated rapid eye movement (REM) sleep behaviour disorder (iRBD), which is a failure of the normal suppression of muscle activity during REM sleep. iRBD is now recognized as a very early symptom of synucleinopathies such as Parkinson's disease. Synucleinopathies are characterized by the aggregation and spread of misfolded α-synuclein through the central nervous system, and associated neuroinflammation. A failure of cerebrospinal fluid drainage can only make this worse, closing off one way to remove aggregated proteins, and increasing inflammation as a consequence.
CSF turnover dysfunction: a hidden early biomarker in iRBD?
Evidence in Alzheimer's disease and other dementias shows that changes in cerebrospinal fluid (CSF) turnover and perivascular spaces (PVS) volume are associated with disease progression through impairment of waste-clearance glymphatic pathways. Volume of CSF, PVS, and drainage structures such as venous sinus are mostly excluded in current MRI studies of premanifest synucleinopathy.
Here, we used MRI to investigate whether modifications in CSF, PVS, and venous sinus volumes occur in 18 prodromal synucleinopathy patients (namely isolated rapid-eye-movement sleep behavior disorder, iRBD) compared to 20 healthy young and 18 elderly controls. Our results demonstrated increased CSF and PVS volumes in iRBD without a matching increase in drainage venous structures, as observed in elderly controls. This suggests increased CSF and PVS fluid stasis, possibly due to impaired CSF filtration, a mechanism that could reduce glymphatic function and exacerbate the neurodegenerative process in iRBD.
A new study associated with Immorta Bio suggests that combining a senolytic vaccine with mesenchymal stem cells might create a synergistic impact. However, the findings rest on acute, artificially induced injury models rather than natural aging [1].
Mesenchymal stem cell (MSC) therapies have largely underperformed in the clinic. MSCs are connective-tissue stem cells that help mostly not by becoming new tissue but by secreting repair-promoting factors. Despite strong preclinical promise, clinical MSC trials in fibrosis, inflammation, and organ failure have shown only modest benefits [2].
One of the reasons may be an unwelcoming environment full of senescent cells, which secrete a mix of inflammatory and tissue-degrading molecules called the senescence-associated secretory phenotype (SASP). Prior work suggests that SASP factors actively suppress stem cell proliferation, differentiation, and survival [3]. In a new study published in the Journal of Translational Medicine and associated with the biotech startup Immorta Bio, the authors suggest a solution: combining MSCs generated from pluripotent stem cells with the company’s proprietary senolytic agent SenoVax.
As evident from its name, SenoVax is a “senolytic vaccine” that primes the immune system against the body’s own senescent cells. Notably, Immorta describes SenoVax in two different ways. In its patent, IND, and press materials, SenoVax is presented as an autologous, personalized cellular immunotherapy: the patient’s own cells are taken via biopsy and driven into accelerated senescence, then used as an antigen source to pulse the patient’s dendritic cells generated ex vivo. The dendritic cells are then reinfused and prime T cells. This is a personalized, work-intensive, and expensive procedure. In the study, however, SenoVax is described as a simpler peptide-based vaccine: peptides derived from senescence-associated proteins and injected subcutaneously along with an immune-triggering adjuvant in the hope that resident dendritic cells will “learn the lesson” in vivo.
The researchers tested the combination in two mouse models of senescence-driven damage, asking whether the combination beats either therapy alone on inflammation, regeneration, organ function, physical performance, and survival. One model involved injecting carbon tetrachloride (CCl₄), a liver toxin, to emulate chronic liver injury and senescence-associated inflammation. The other one was based on injecting low-dose doxorubicin, a chemotherapy drug that drives cells into senescence. In each model, induced mice were split into four arms: untreated control, SenoVax alone, MSCs alone, or the combination.
The team then measured four inflammatory/SASP markers – IL-11, IL-23, IL-6, and YKL-40 – in the liver injury model. All four fell below the injured baseline in every treated arm, and the combination lowered each one the most, suggesting that both agents dampen SASP signaling and that combining them produces the largest effect. Importantly, these factors are less senescence-specific than p16, p21, or SA-β-gal, so the senolytic mechanism is rather inferred than shown. Conversely, the regeneration markers Klotho, FGF-2, VEGF, and GDF-11 rose above the injured baseline, while the liver-damage enzymes AST and ALT fell; both of these shifts pointed toward improvement. In each case, the combination moved furthest, supporting the idea that clearing SASP takes the brakes off regeneration.
To show the pattern isn’t specific to chemical liver injury, the researchers then repeated the SASP and the regenerative markers panels in the doxorubicin-induced “accelerated aging” model. The results were similar: most positive with the combination.
To test physical function in the “accelerated aging” model, the team used the “T-climbing” test, which times how long a mouse takes to climb down a vertical pole – a standard motor-coordination and strength assay. The combination improved climbing performance by roughly 65%. However, this claim, made in the Discussion session, is not supported by the correspondent figure, which only contains bars for single interventions, not for the combination. Numbers for monotherapies do not appear in the paper.
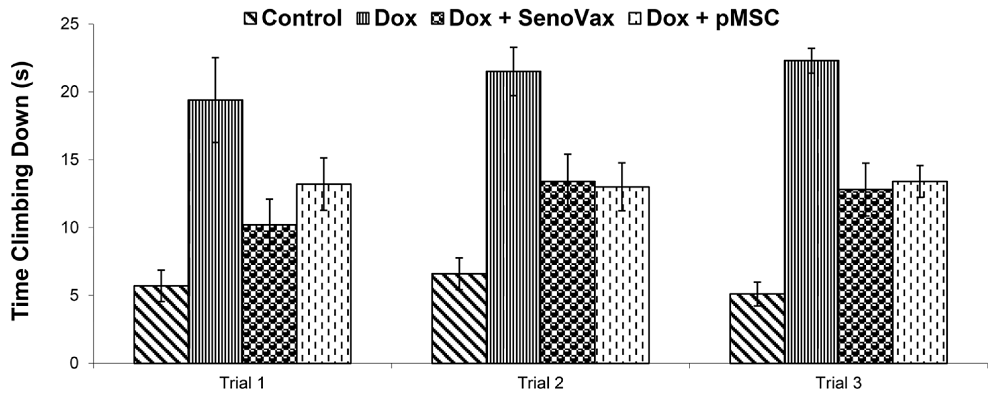
In the capstone experiment, which tested survival, mice received doxorubicin until death or a humane endpoint. The combination again gave the best results: about 50% of the animals were alive at Day 35 and 20% at Day 40, versus complete mortality by Day 30 in untreated doxorubicin controls. Monotherapies extended median survival only modestly, to about Day 35.
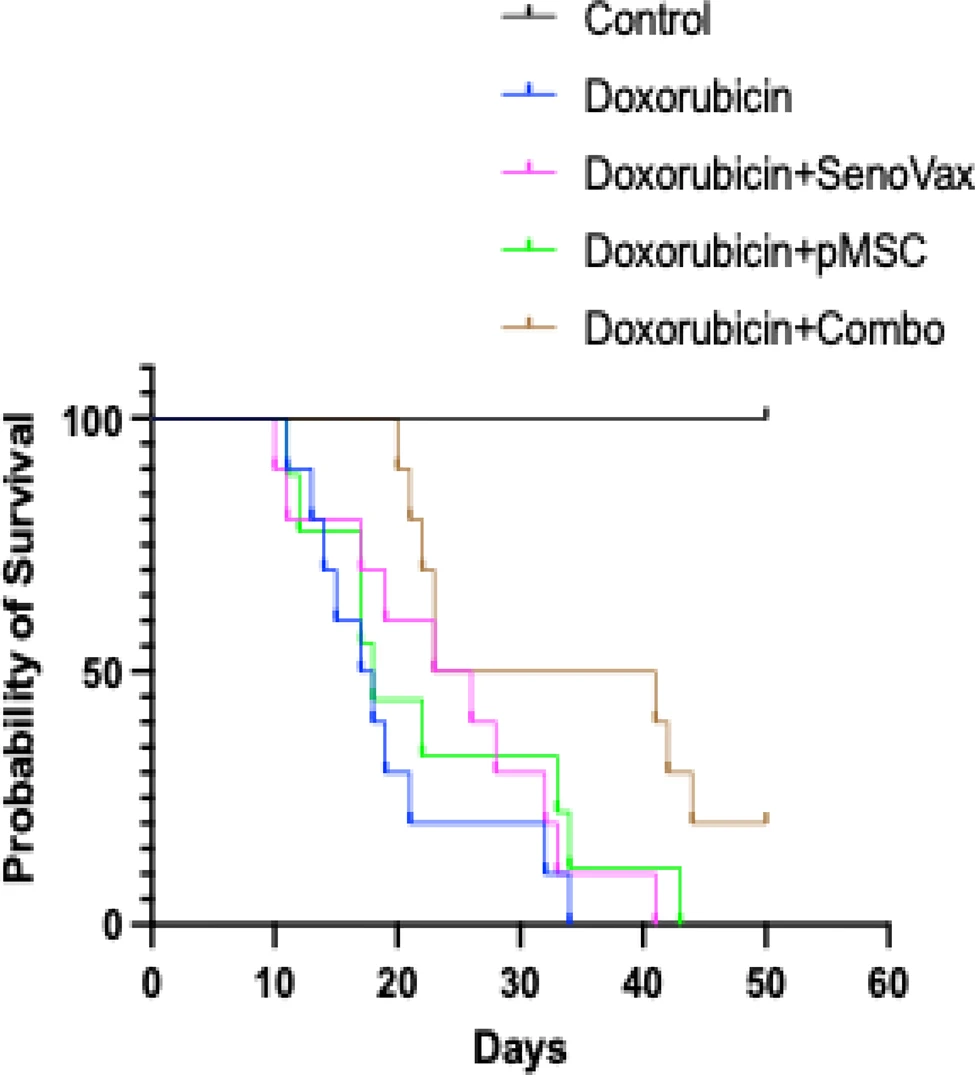
While the accompanying press release touts a 73% increase in mean survival and ~84% extension of median lifespan compared with untreated controls in validated murine aging models, the extremely short lifespan puts it more into the “acute toxin-related damage” territory, as opposed to accelerated aging, much less natural aging.
Despite the several drawbacks and quirks, the study lends certain support to the intriguing concept behind Immorta Bio: using senolytics to create an auspicious niche for MSCs to work their magic. Hopefully, the company will keep developing this concept further.
[1] Ichim, T. E., Markov, N., Lopes, G., Pascual, K. A., Evans, A., Reznik, R., … & Reznik, B. N. (2026). Synergistic senolytic–regenerative therapy significantly extends healthspan and lifespan Journal of Translational Medicine, 24(1), 745.
[2] Levy, O., Kuai, R., Siren, E. M., Bhere, D., Milton, Y., Nissar, N., … & Karp, J. M. (2020). Shattering barriers toward clinically meaningful MSC therapies. Science advances, 6(30), eaba6884.
[3] Moiseeva, V., Cisneros, A., Sica, V., Deryagin, O., Lai, Y., Jung, S., … & Muñoz-Cánoves, P. (2023). Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature, 613(7942), 169-178.
Vaccinations can produce a lasting effect known as trained immunity, altering the behavior of the innate immune system and resulting in both a reduction in the chronic inflammation of aging and a more effective immune response to unrelated infectious agents. Arguably the largest body of research into trained immunity involves the BCG vaccine for tuberculosis, widely used outside the United States. A lesser body of work examines the trained immunity effects of a small number of other vaccines that are more commonly provided to adults rather than children. For most vaccines, there is no human data, and trained immunity itself is well documented, but not well understood in detail. Here, researchers report on a small trial to assess the effects of BCG vaccination on older people with and without Alzheimer's disease, to see whether the changes produced by trained immunity are sizable enough to justify further trials and widespread use.
Immune aging may contribute to Alzheimer's disease. Bacillus Calmette-Guérin (BCG), a vaccine known to induce trained immunity, has been linked to reduced Alzheimer's risk in prior studies. However, whether trained immunity can be observed in the human central nervous system remains unclear. We conducted two related one-year, open-label clinical trials in adults aged 55 years or older (n = 12 without Alzheimer's-related pathology; n = 11 with Alzheimer's-related pathology) recruited at a single center. Participants received two intradermal BCG vaccinations one month apart.
We show that BCG induces persistent, trained immunity-like changes in immune cells in cerebrospinal fluid, including enhanced innate responsiveness and associated transcriptional programs. These responses differ from blood, suggesting compartment-specific immune imprinting. In participants without Alzheimer's-related changes, these immune shifts are accompanied by decreased amyloid-β levels in cerebrospinal fluid and increased levels in blood. BCG was well tolerated, with no unexpected safety signals observed. This approach may represent an early neurodegenerative intervention strategy, although larger controlled studies are needed to confirm these observations.
Link: https://doi.org/10.1038/s43856-026-01691-7
Senescent cells are involved in tissue regeneration. Cells enter a senescent state following injury, and in the usual course of events assist in the intricate coordination between immune cells, stem cells, and other cell types that is required to regrow tissue. These senescent cells are then cleared by the immune system. With age, in poorly regenerative tissues, or in severe or persistent injuries, the presence of senescent cells following injury can become excessive and maladaptive. The signaling produced by senescent cells lasts too long, the cells are not cleared, and it causes further harm. As researchers here note, this is what happens following a heart attack, suggesting that there is likely some timing for the delivery of senolytic therapies to selectively destroy the cells become senescent immediately following the ischemic injury of a heart attack that could improve patient outcomes.
Currently, the primary causes of death following myocardial infarction include sudden cardiac death, malignant arrhythmias, and acute heart failure, all resulting from myocardial necrosis caused by coronary artery occlusion. Cellular senescence refers to the permanent arrest of cell proliferation in response to stress stimuli; it serves as a crucial tumor defense mechanism and is closely associated with tissue aging and chronic inflammation. The senescence-associated secretory phenotype (SASP) is one of the most characteristic features of senescent cells. Cardiac cells develop a SASP and secrete SASP factors in response to stimuli such as oxidative stress, DNA damage, and hypoxia, playing a key role in immune regulation and tissue repair following myocardial infarction.
The SASP exhibits marked spatiotemporal heterogeneity following myocardial infarction: during the acute phase, it contributes to inflammatory amplification and immune cell recruitment; during the subacute phase, it is involved in inflammation resolution, matrix remodeling, and scar formation; and during the chronic phase, it promotes chronic inflammation, paracrine senescence, pathological fibrosis, and cardiac dysfunction. Spatially, the SASP influences scar stabilization in the infarct zone, inflammation-electrophysiological coupling in the border zone, and compensatory remodeling in the distal region. The sustained expression of the SASP is a major driver of adverse ventricular remodeling and excessive fibrosis following myocardial infarction.
Therefore, targeting senescent cells and persistent, pathological SASP represents a highly promising therapeutic strategy in the field of cardiovascular regenerative medicine. This review will discuss senescence in different cell types following myocardial infarction, the spatiotemporal heterogeneity of the immune response mediated by the SASP after myocardial infarction, and the immune cells regulated by the SASP.
Link: https://doi.org/10.3389/fimmu.2026.1850084
0 members, 2 guests, 0 anonymous users


Community Forum Software by IP.Board
Licensed to: ImmInst.org


























