I really wanted this post to be comprehensive and conclusive. I can feel the shape of the answer in my head, but there are so many puzzle pieces I can’t yet fully complete it. But it is still pretty good. In the places where I’ve indicated I am missing something, if you can help, please do. I really think that aging is close to being understood. I have never been more optimistic that aging will be solved, and soon. Now I need to get this out before I burst,.....
Please stick with me whilst I try and lay out all the puzzle pieces.
Let me summarise quickly:
- Oxidant stress decreases TET enzymes, post #617 (above)
- TET enzymes keep GDF11 promoter ‘clean’ of methylation so it can be expressed, post #424
- Methylation (in women’s skin) of the oxytocin receptor contributes to skin aging, post #473
- High ROS environment decreases TET activity and methylates klotho promoters, leading to kidney disease, post #486
- Part jigsaw piece - Vital mitochondrial energy production is turned down via age related methylation changes! Remember that paper back in 2015 that showed simple glycine could reverse age related mitochondrial defects? Well the relevant nuclear gene in humans, SMHT2, is important for methylation (via making glycine for SAM) but also in making new mitochondrial DNA (via making N-formylmethionine, which is the starting amino acid in mitos and bacteria, not just plain methionine like in human DNA). Hence the SHMT2 gene is vital in every cell’s energy production and it is downregulated with age. I have not yet found a link to exactly how this happens, but we do know that Yamanaka factors reverse SHMT2 silencing and we know cell reprogramming doesn’t work without the TETs (as shown by David Sinclair). Much of what I know is from the following paper https://www.nature.c...cles/srep10434
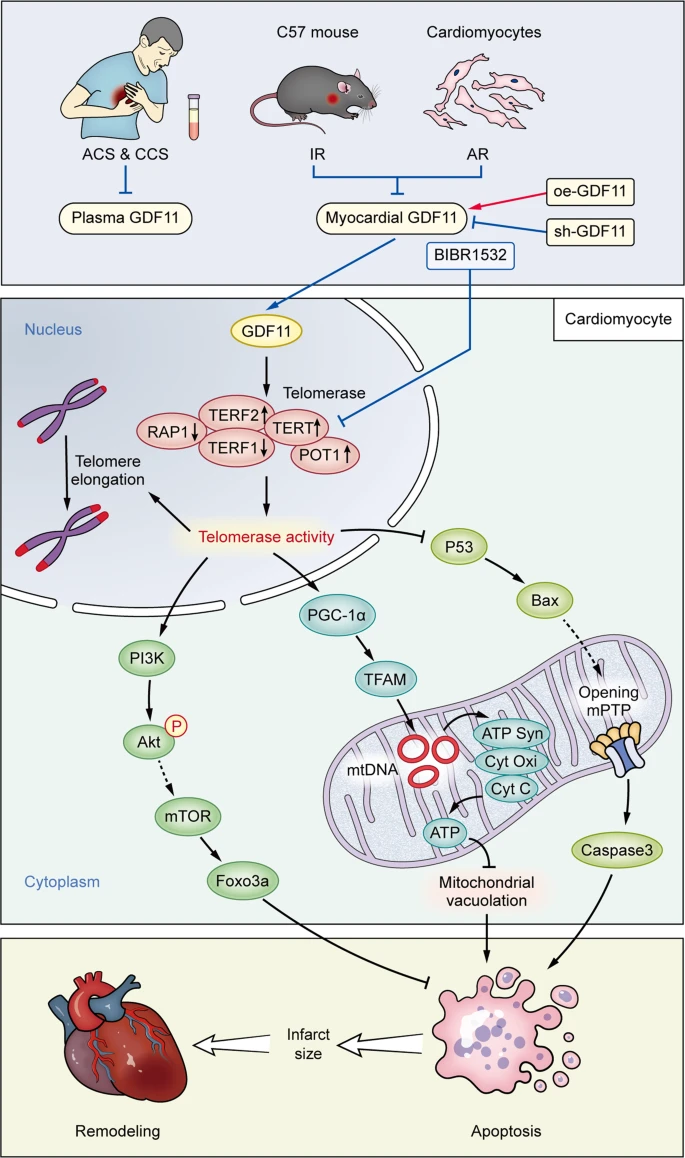
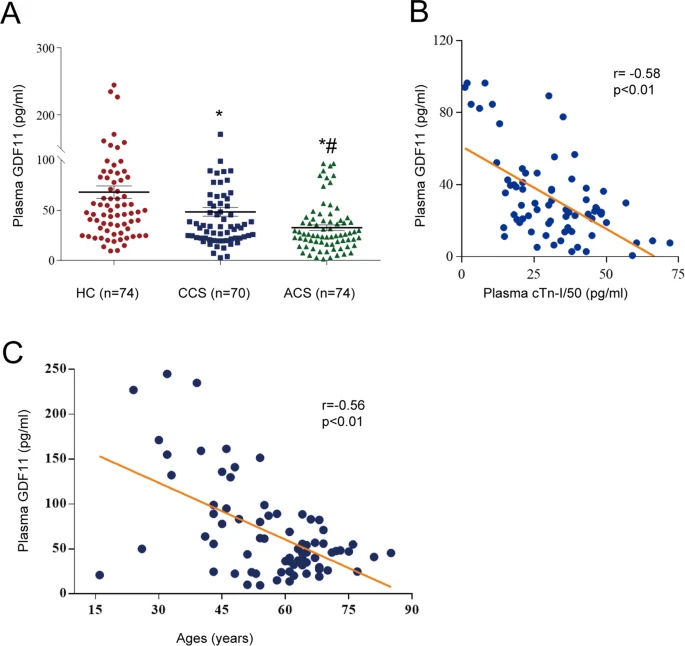
The above posts and references all show that age related methylation (which we know from the Horvath reference in the last post is much more prevalent in aging than demethylation) is causing the inactivation of important genes. Most interesting is GDF11, which is pro-differentiation. On that subject I have always been worried that molecules that cause increased differentiation, like retinol in skin cells or stem cell stimulants like AFA or C60, or GDF11 (even though it also increases telomerase), could cause stem cell exhaustion. But there has been growing evidence that much of aging is actually the reverse problem; stem cells are still there but they aren’t differentiating.
In a recent experiment they found that Loss of Dnmt3a (a de novo methyltransferase) Immortalizes Hematopoietic Stem Cells In Vivo, but that they would no longer differentiate! https://www.scienced...211124718303541
These HSCs could proliferate forever and never lost telomeres
Dnmt3aKO HSCs showed no erosion of telomere length
, but never produced any blood cells for the poor mice hosting them
Also from https://www.nature.c...rticles/ncb1386 , Blasco and friends showed,
Mouse embryonic stem (ES) cells genetically deficient for DNMT1, or both DNMT3a and DNMT3b have dramatically elongated telomeres compared with wild-type controls. Mammalian telomere repeats (TTAGGG) lack the canonical CpG methylation site. However, we demonstrate that mouse subtelomeric regions are heavily methylated, and that this modification is decreased in DNMT-deficient cells.
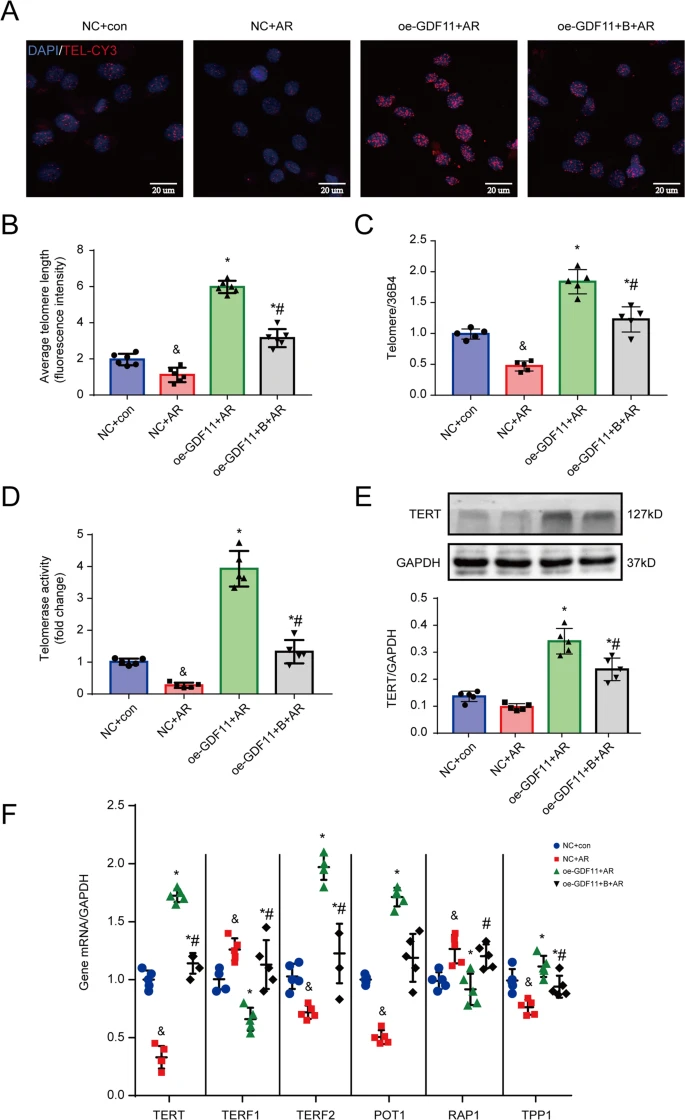
So telomerase is under methylation control.
We also see an increase in de novo methyltransferases with age, see; https://pubmed.ncbi....h.gov/17929180/. I don’t know if the demethylators (TETs) failing with age is related to the de novo methylators increasing with age., but I’m throwing it in here. Note cocoa decreases DNMTs (https://pubmed.ncbi.nlm.nih.gov/23840361/).
Moving back to telomeres, I spent a lot of time recently looking at how in both human and mice cells, differentiation downregulates telomerase production, see post #588. It seems likely that this is accomplished via DNMTs. But more importantly this might be the ‘selection pressure’ causing the whole of aging. My Selfish Cell theory of aging says that stem cells that differentiate die, whilst those that stay as stem cells live; see Post #122 and Post #130. You can see that even back then, I was on the right path.
Stress causes differentiation of progenitor cells or harder work in somatic cells . This is an intentional response to support tissues. But it will over time select for cells that are resistant to stress AND resistant to responding to that stress. With age ever greater stress is required to renew tissues. Although this gives us some confidence that exercise, CR, IF, autophagy inducers, etc. will slow aging this has a practical limit. Other treatment options are required.
Allow me a metaphor: The body survives only because of the sacrifice of cells being willing to differentiate and potentially die, rather like a soldier being willing to die for his country. Once the country only has lazy, selfish people who put their needs above that of the country, it's days are surely numbered. But in times of bodily stress (like War for a country), the best tragically die first. Life is constant stress on the body that requires differentiation of cells. But differentiation turns down telomerase. We are literally training the cells of our body to do what those selfish HSCs cells with no DNMT3a did by human design - proliferate forever and refuse to differentiate. We are aging because we are slowly turning back into stem cells.
In post #476, I talked about how ‘aging is cancer’. This was a pretty ballsy statement. But the referenced study ‘DNA Methylation Patterns Separate Senescence from Transformation Potential and Indicate Cancer Risk’ https://www.scienced...535610818300084 showed that the cells of aging humans are more commonly like cancer cells than senescent cells, i.e. they bore methylation patterns that looked more like stem cells than cellular senescence. Going back to GDF11 for a moment, bear in mind that Steve Perry has shown GDF11 is robustly anti-cancer for some cancer types in dogs. I wouldn’t be surprised if GDF11 is generally anti cancer because it encourages proper differentiation of pre-cancerous or actual cancerous cells.
I speculate that the body takes more extreme measures to force stem cells to differentiate and replenish tissues as we age, and this might be behind the rise in the destructive hormones Jeff Bowles talks about so much (LH, FSH, etc). But even though this might be successful in causing continued differentiation, the cells produced are not phenotypically normal, and bear a pre-cancerous phenotype as shown in the above study. A skin cell is not fully a skin cell, a liver cell is not exactly a liver cell. I think of elevating growth signalling to replenish tissues from an unwilling stem cell pool to be like driving with both the accelerator and brake pedal pressed down (or driving with the handbrake on).
There is also the possibility that pro-oxidant like hormones (LH, FSH) actually cause a downregulation in the TETs via ROS, and this would fit with growth signals like MTOR being necessarily for growth and development from a child but later causing unwanted methylation of important differentiation genes, as we’ve already discussed. This is an area where I don’t have all the jigsaw pieces yet. It does link nicely with anti-oxidant hormones like melatonin being anti aging (see Post #472), whilst also downregulating sex drive, potentially delaying menopause etc (more Jeff Bowles stuff). At the moment my stance is the hormone elevation happens too late to be the initiator of aging, but it might deliver the coup de grace. Or it might be a gradually increasing feedback loop as the stem cell pool becomes dominated with ‘selfish cells’; it starts rising gradually and this accelerates until you literally destroy the tissues producing the hormones.
An interesting consequence of growth signals forcing reticent stem cells to differentiate, is that this might cause the preservation of telomere length in the very old. As partially differentiated cells predominate, some telomerase expression may be preserved. See: https://www.ncbi.nlm...les/PMC4634197/
In this kind of study you see telomere length falling with age (it is only a cross sectional, so they aren’t the same people) and then rise in the centenarians that survive the longest. They then say telomeres are not important for aging. I always put this down to survivor bias, with those with long telomeres being those that survive to very old age, but maybe there is more to it and the very old have longer telomeres because telomerase is no longer being shut down in their partially-differentiated cells. Typically long telomeres and active telomerase are blamed for cancer by lazy researchers. But my work here shows another possibility, that cells are becoming more cancerous because of selection pressure on cells to be selfish, and this naturally preserves telomere function because of the lack of methylation-mediated downregulation that comes with proper differentiation.
That’s been a long post!
Conclusions:
- The body needs constant replenishment and this is accomplished by differentiation from the stem cell pool into vital specialist roles (skin, liver, blood, vessel lining, etc.)
- Differentiation can only occur if important developmental genes are kept demethylated. Elevated ROS can impede the demethylases (TETs) and cause these genes to be methylated, blocking differentiation and causing the cells that can be made to differentiate to have a stemlike/pre-cancerous phenotype.
- Differentiation also causes downregulation or complete abolishment of telomerase, therefore eventual death via ROS/telomere attrition/both for that cell line (this downregulation of telomerase is accomplished by de novo methyltransferases DNMTs. Blocking DNMT3a caused blood stem cells to become immortal but never produce blood cells). DNMTs go up with age, possibly as a defence mechanism against rising pre-cancerous cell numbers. Longer telomeres in the very old might be evidence of telomerase not being properly turned off in spite of this.
- The source of the rising ROS that disabled the TETs and stopped differentiation in the first place might be the pro-oxidant hormones (LH, FSH) required for growth from a child to an adult. (There are numerous other possible triggers that could start this off, which is why aging is so robust).
- Because differentiation is basically a telomerase-blocking death sentence for cells (but a lifesaver for us) as time goes on ‘Selfish’ stem cells will come to dominate the reserve pool - these cells require stronger and stronger growth signalling to differentiate, and this might be behind destructive sex hormones rising in late middle age.
Personal Epilogue
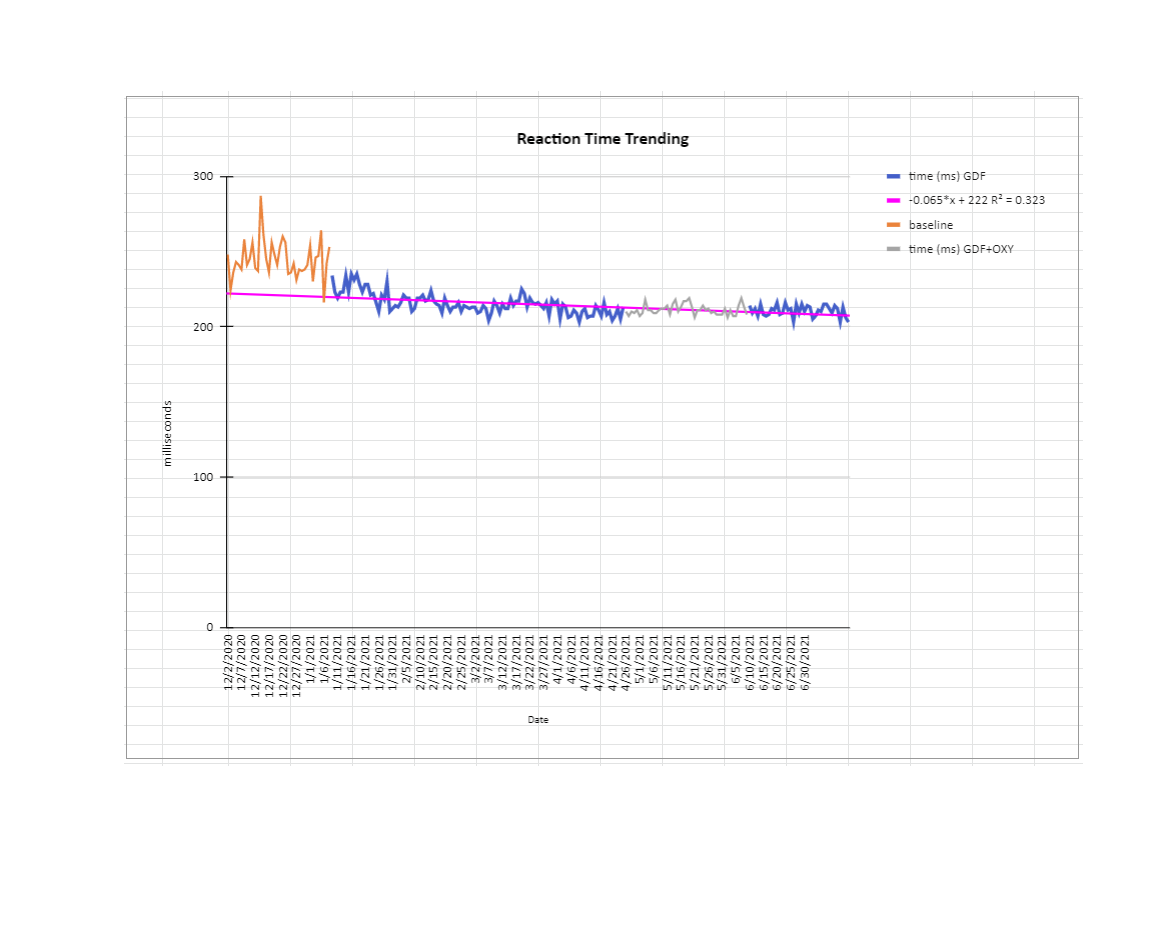
I haven’t got time to write a whole lot on ‘what should we do about this’ given everything I’ve said. For now know I haven’t changed my view on telomerase activators (I use TAM818, also Vit D (+K2)in the winter, I also take B6/9 with it), anti-oxidants (telomerase protects mitochondria and lowers ROS, melatonin/epitalon is probably the best anti-oxidant; but I also like Vit C for demethylation and ROS, and beta-carotene plus Zinc for multiple reasons) or AKG (surely a godsend discovery for upregulating TETs, but requires cycling ), GDF11 (in small amounts is very useful for noticeable de-aging, particularly with AKG) and mTOR inhibitors to temporarily decrease proliferation and give telomeres a chance to lengthen (I mainly use coffee, occasionally rapamycin/everolimus - but I get side effects really easily); There are lots of other possible interventions like klotho, oxytocin, (good) sex hormones (that fall with age), stem cell stimulants like C60 and AFA. I’ve tried oxytocin and it didn’t seem additive with other things I’m doing. AFA works but can cause fatigue. I think C60 could be a winner, but I need to find a manufacturer I trust. Klotho is definitely high on my list. I’ve haven’t tried pregnenolone/DHEA etc.
A personal note: mentally I feel as good, or better, than I ever have. Maybe the closest I can recall to this is being a rather acerbic 16 year old with a ruthless intellect intent on criticising the world. Luckily the return of my mental acuity has not turned me back into a teenager. Physically I can work out every night if I want to now. My skin still looks older than I’d like, though considerably younger than many of my peers. My hair is full and dark. I have a little grey in my beard. I wonder whether the ‘damage’ theories of aging will still be relevant at some point - even if damage is due to aging and not the cause of aging, it is still damage and will not necessarily all disappear once full youthful homeostasis in the body is restored. I expect this will mainly be a problem for those who are already very old and consequently have a lot of unrepaired damage.
Edited by QuestforLife, 15 July 2021 - 10:52 AM.