Finding the culprit: the hormones required for sexual maturity may be the trigger that starts aging via downregulation of TET2

The above figure by Blagosklonny is highly relevant to our discussion of hormones, (methylation) and aging.
He describes the figure as follows:
Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives
by Blagosklonny
doi: 10.18632/aging.100149
The menstrual cycle is regulated by interplay of negative and positive feedback loops. The hypothalamus stimulates the pituitary gland to secrete Follicle-Stimulating Hormone (FSH), which in turn stimulates follicles in the ovaries. Follicles maturate and secrete estrogens. Estrogens inhibit the hypothalamus, decreasing secretion of FSH (a negative feedback loop). In turn, FSH stimulates ovarian follicles, which produce estrogens, which in turn inhibit FSH production. Also, estrogens stimulate secretion of Lutenizing Hormone (LH). LH in turn causes ovulation. So for the normal menstrual cycle, the hypothalamus should have a narrow range of sensitivity to estrogens. Both too high and too low sensitivities are not compatible with menstrual cycles. In comparison, regulation of reproduction in men is simpler. There is a gradual decrease in fertility in men too (analogous to pre-menopause), although this usually does not result in testicular failure during a man's lifetime.
You could argue, and I would, that the real cause of aging in this scenario is the limited number of activatable follicles and egg cells therein. And this links aging back to the telomeres of oocytes, which are finite. Indeed, we see numerous issues in older mother relate to short telomeres in eggs (can discuss another time as it is a big subject).
But as we know the same thing occurs with men (albeit slower), with supposedly immortal spermatozoa. I have discussed this before, in post #474, and I won’t go any further into it here, other than to say aging also affects the male reproduction system in a way compatible with the idea that sperm are failing to differentiate properly.
From Post #474: Spermatogonial stem cells are already immortal, but another paper [10] shows that a similar process is occurring (although there is no evidence at this time that this is mediated by increased methylation); with age sperm cell progenitors reduce mitochondria, increase glycolysis and self-renew at the cost of differentiation and testicular atrophy (ouch!). [10] source: www.pnas.org/cgi/doi/10.1073/pnas.1904980116
How does this all link up to hormones and aging? It is possible that the feedback mechanisms that time and trigger puberty also lead to aging, but on a less well timed basis, as suggested by Blagosklonny with menopause. Blagoskonny clearly explains that an initial small amount of LH and FSH from the hypothalamus and the pituitary gland activate a small amount of estrogen from the follicles, which is sufficient to shut down LH and FSH. Later the hypothalamus and the pituitary get resistant to estrogen and the loop escalates until there is sufficient LH/FSH to release an egg (and menarche begins). The large quantity of estrogen released with the egg knocks LH/FSH right down and the build-up begins again, cumulating a month later with another egg being released. This continues until the eggs all run out, and even though LH/FSH stays high from then on, no eggs are released and the system is broken.
Note: there are reports melatonin and NAD+ boosters can restart menarche; this fits with my previous post linking increased oxidative stress to methylation and blocked differentiation (in this case oocytes). There might be some egg cells still remaining after menopause, but they can’t be made to mature whilst oxidative stress is high. I also do believe there is a hard limit here, but I’ll discuss that on request.
Could the aging of the female reproductive system be extended to the whole organism? I believe so. The following paper offers a tantalising hint:
An epigenetic switch repressing Tet1 in gonadotropes activates the reproductive axis
Source: https://www.pnas.org...nt/114/38/10131
We present an epigenetic switch in the central control of reproduction as a truncated TET1, expressed in proliferating gonadotrope-precursor cells, which inhibits Lhb (=luteinizing hormone) expression and so must be repressed for reproductive development. Expression of this TET1 isoform is regulated by cis-elements mediating effects of gonadal steroids and PKA, and also a potentially methylated distal enhancer. As this isoform appears more common than the canonical TET1 in other differentiated tissues, our study has broader functional implications outside of the reproductive axis. Furthermore, our findings support the idea that distinct genomic regions are used at different developmental stages or in different tissues, and that a particular sequence can be part of the primary transcript in some tissues or an enhancer RNA in others.
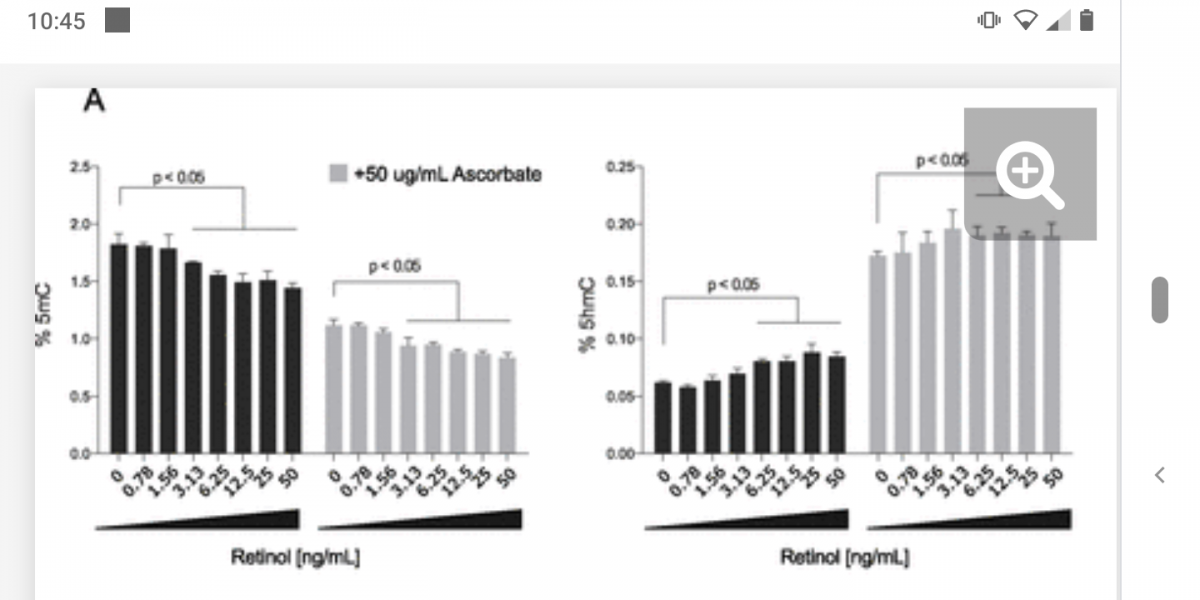
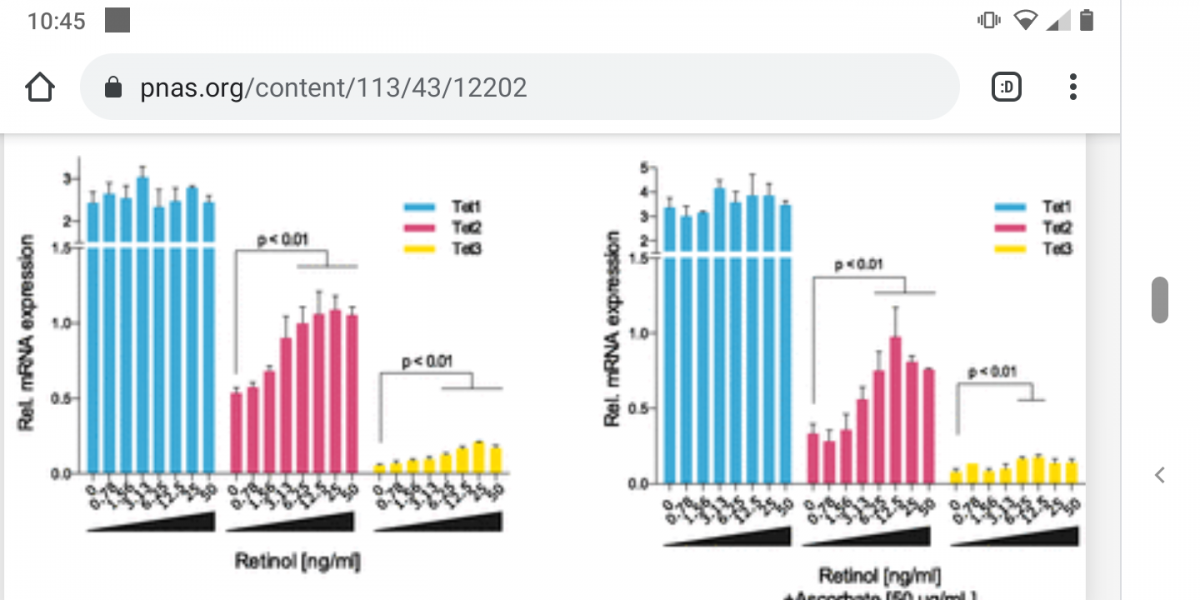
The paper gives more detail: reproductive cells upregulate LH as TET1 is downregulated (as an intentional programmed part of maturity of reproductive cells; note TET1 is normally only expressed in embryonic cells, TET2/3 is expressed other tissues). But they also show other non-reproductive differentiated cells have an isoform of TET1 expressed (an alternatively spliced version), which keeps LH suppressed in those cells. This can be overcome by a range of sex hormones or by methylation of TET2, which keeps the TET1 promoter turned on by removing its methylation.
It all comes back to those pesky TETs! Methylation of TET2 not only messes up things like GDF11 expression and oxytocin receptors and (possibly) mitochondrial health, but also causes random tissues in the body to turn on LH. We know LH is an oxidative hormone, and this will cause further downregulation of TET2 and ratchet the feedback loop to more methylation and more LH and less TET2. Where does this all start? Given we need LH (and FSH if you’re female) to reach sexual maturity, it is possible that this LH/FSH ‘leaks’ and gradually turns up methylation across all body tissues. And it is not as if we can do without these hormones; without FSH/LH women would release no eggs, and presumably (I need to look into this (*)) without LH men would not mature sperm cells. The signal to reproduce is also the signal for aging (on a much slower timescale) with women getting a surge of aging with their period each month, turning up to a constant surge of aging after menopause, and men getting a constant aging signal, which likewise ratchets up with LH in late middle age.
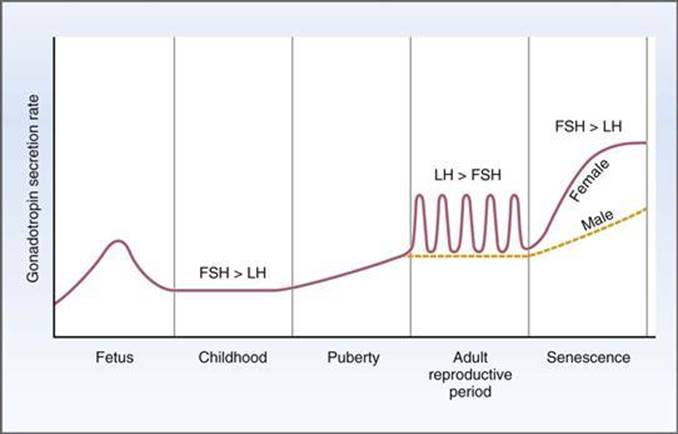
Source: https://doctorlib.in...ology-2/91.html
Note how there is a spike in the gonadotrophins in the fetus; perhaps related to the downregulation of TET1 and the creation of sex organs. The next time the hormones are that high triggers sexual maturity, thereafter levels are constant (averaging the monthly female cycle), until late life increases them yet further.
(*) Early research on male fertility initiation and aging
Gonaldotropins are required for spermarche (start of sperm production), see https://pubmed.ncbi....ih.gov/2492750/
But rising luteinizing hormone also causes loss of quality of sperm in later life, see;
https://bmcurol.biom...894-020-00674-7
Edited by QuestforLife, 19 July 2021 - 12:11 PM.