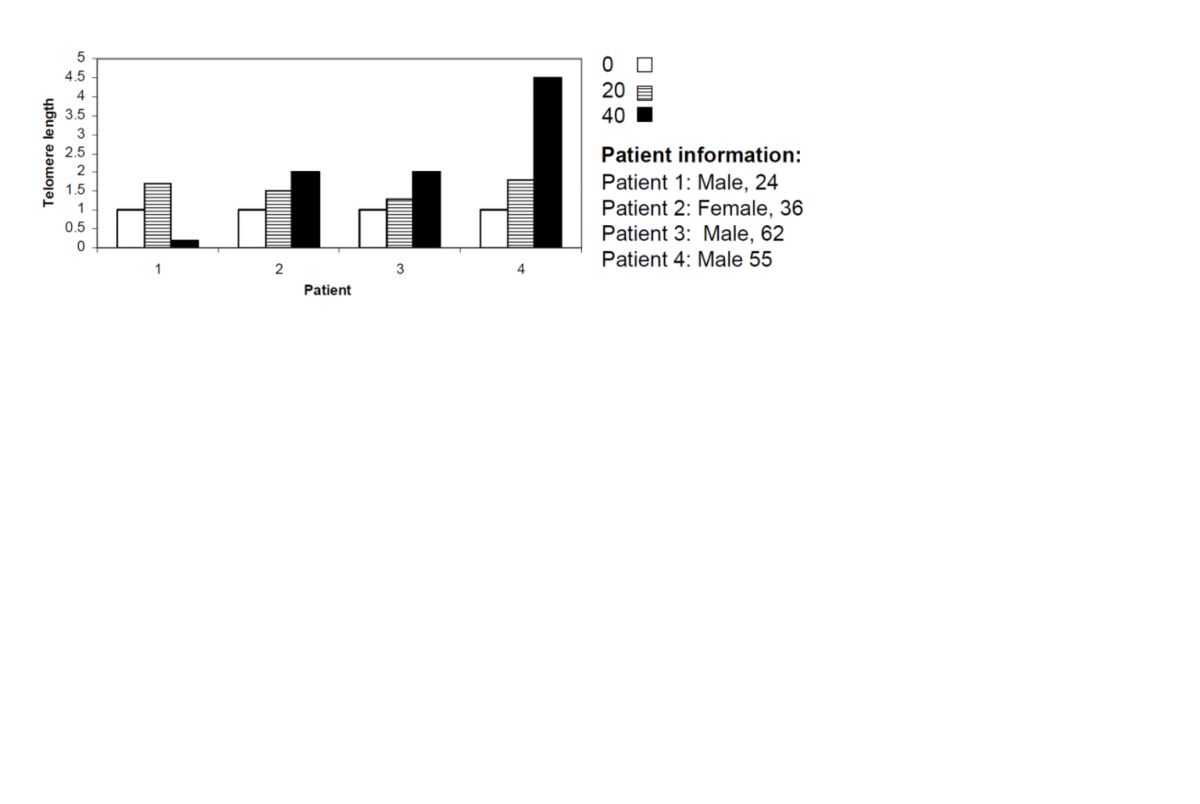
Has the Telomere Theory of Aging been proven?
A long while ago I discussed this Blasco paper, where amongst other things they demonstrated that kidney cells with short telomeres undergo the EMT (epithelial to mesenchymal transition). This was reversed (MET) by extending telomeres. I speculated at the time of the importance for cancer, given the main source of cancer in humans is the epithelium.
I've also discussed before how with age cancer-like cells become more and more common, to the extend that I claimed 'cancer is aging.'
And as everyone knows by now, I'm a big fan of the telomere theory of aging - I think it's the lynchpin of all aging - and I've also never agreed that telomere shortening is all about senescent cells; like Michael Fossel I believe aging is analogue not digital, and that as telomeres get shorter cells become progressively more dysfunctional. It is not a sudden, all or nothing, distinction.
With that lengthy introduction out of the way…
Novel insights from a multiomics dissection of the Hayflick limit
https://elifescience.../articles/70283
This is a very important paper that recreated the original Hayflick experiments of passaging lung fibroblasts to replicative senescence, examining with a fine tooth comb changes in gene expression over time, and importantly controlling for cell density as well as contrasting wld type cells with the parallel changes in telomerase immortalised fibroblasts, as well as cells made senescent through radiation. As you read my highlights from the paper, you'll see what an important work it is, and why it's justified such a lengthy introduction.
I give you some choice quotes.
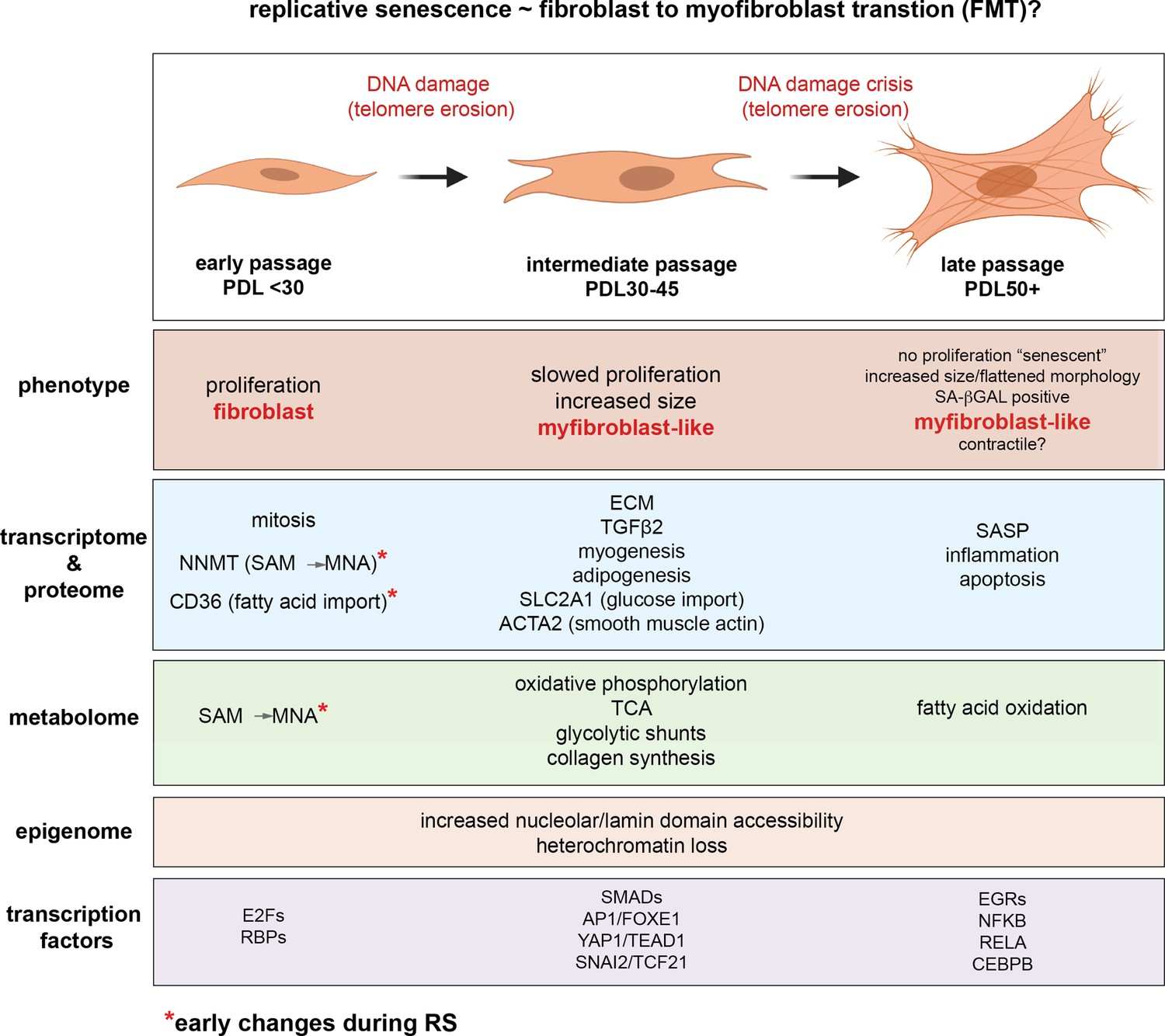
…the time resolution of our experiment coupled with single-cell trajectory analysis reveals that senescence is a gradual process that shares transcriptional, proteomic, and metabolomic features with epithelial-mesenchymal transition (EMT). Second, our metabolomic data identifies Nicotinamide N-methyltransferase (NNMT) activity as a potential initiating event in replicative senescence dependent loss of epigenetic silencing. Third, we show that these genomic regions that exhibit increased accessibility with increasing cellular age are concentrated in nucleolar/lamin associated domains and correspond with observed changes in the replicative senescence transcriptome...as expected, gene expression in the hTERT-immortalized WI-38 cells remained largely stable…the gene set for epithelial to mesenchymal transition (EMT) was significantly enriched in genes that increased with replicative senescence as opposed to radiation induced senescence…the EMT gene set is enriched early and robustly during replicative senescence…Interestingly, the vast majority of gene expression changes evident by PDL 50 begin to manifest at much earlier PDLs.
They carried out single-cell RNA experiments to see if the changes they were seeing was due to the changing mix of cells (some senescent, some not) or due to changes across all cells simultaneously. Remarkably they found the latter was true: the vast bulk of cells are moving together towards senescence. I cannot stress this last point enough. In their words.
Together, these results argue in favor of a gradual model of replicative senescence wherein cells ramp up expression of the replicative senescence program with increasing PDL even when still proliferative... Importantly, this observation suggests that aspects of cellular senescence are present in non-senescent cells. This result raises the intriguing possibility that the reported pathological features of senescent or senescent-like cells in vivo might also manifest in cells that are not classically senescent.'
From the proteomics data.
Multiple enriched sets pointed to replicative senescence-dependent shifts in cellular energy utilization…We observed PDL-dependent increase in proteins from all categories except for mitochondrial assembly and structures. These results are consistent with altered mitochondrial function…From these data, it is clear that WI-38 cells approaching replicative senescence undergo drastic shifts in metabolism. Specifically, we observed increased glucose utilization in glycolytic shunts coupled with an increase in fatty acid import and oxidation. It is possible that the senescent cells are switching to fatty acid oxidation to fuel increased TCA cycling and oxidative phosphorylation as glucose is diverted to macromolecule production….Likewise, consensus metabolomic findings in EMT models report increased TCA cycle products, altered lipid metabolism, and activated hexosamine pathway. These data provide functional metabolic evidence supporting a connection between replicative senescence, metabolic rewiring, and the increased expression of EMT-related genes.
The point about increased fatty acid oxidation is the opposite of that seen in the muscles of the elderly. This likely reflects difference consequences of aging in mitotic and post mitotic cells. But it matches what we know about cancer-primed cells increasing with age and shows this is due to telomere shortening. Moving on…
In review of our replicative senescence data for the metabolic and proteomic components of the NAD and methionine pathways, we found that one of the largest and earliest changes for any protein (or transcript) was the increased expression of Nicotinamide N-methyltransferase (NNMT)…DNA and histone methylation require abundant levels of the universal methyl donor S-adenosyl methionine (SAM). NNMT not only depletes SAM by catalyzing the removal of its methyl group, it does so by fusing the methyl group to the NAD+ precursor nicotinamide (NAM) resulting in the generation of methyl nicotinamide (MNA). Thus, NNMT effectively acts as a sink for two of the primary metabolites a cell requires for silencing chromatin and regulating gene expression…High NNMT expression has been implicated in SAM depletion in embryonic stem cells and cancer-associated fibroblasts (CAFs). In both cases, the functional consequences were similar; DNA hypomethylation and decreased capacity to form or maintain silenced loci and heterochromatin. The important point is that the loss of silencing is actively promoted through NNMT activity. One intriguing hypothesis is that increased NNMT activity with replicative senescence promotes loss of silencing and heterochromatin via SAM depletion.
In other words, NNMT upregulation depletes both NAD+ and SAM, affecting both acetylation and methylation of the genome. The chromatin areas that were most affected were associated with the lamina and nucleolus; these were the areas where the drop in SAM and NAD+ most increased chromatin accessibility. So what genes were most upregulated by these changes in the genome accessibility?
A large portion of the transcription factors exhibiting replicative senescence specificity belong to four categories: inflammation transcription factors (NFKB, CEBPB), AP1 subunits (JUN,JUND,FOSL2), YAP1-TEAD1 components (TEAD1/4, YAP1, WWTR1), and EMT transcription factors (SNAI2, TCF21)...'Collectively, these analyses uncover a common theme amongst putative regulatory transcription factors; Hippo signaling (YAP1/TEAD1), EMT transcription factors, and TGF-β signaling (SNAI2, SMAD activity). SNAI2 has been shown to work in tandem with the YAP1/TEAD1 complex and these pathways often work towards similar biological ends. Together, these transcription factors are reported as highly involved with proliferation, EMT, ECM production, fibrosis, and apoptosis avoidance…Collectively, these results present a possible order of operations for replicative senescence progression that highlights an initial cessation of active mitotic cycling, followed by an epigenetic shift that precedes a strong EMT/TGF-β signal before segueing into a pro-inflammatory secretory state…Given the repeated observations linking replicative senescence with EMT, TGF-β and YAP1/TEAD1 activity, we considered the possibility that these processes are connected through the fibroblast to myofibroblast transition (FMT), a subtype of EMT in which stressed or injured fibroblasts differentiate into myofibroblasts. Upon receiving cues mediated by injury or stress (e.g. activated TGF-β), fibroblasts can trans-differentiate into myofibroblasts, whose functions as ‘professional repair cells’ include increased proliferation, migration, apoptosis avoidance, cell and tissue contraction, and ECM/collagen deposition to promote tissue repair and wound closure…Previous work has demonstrated that there exists mechanistic and functional association between telomerase inhibition, senescence, and myofibroblasts. Senescence is an integral part of the wound healing processes; upon injury resolution, activation of a senescence-like phenotype prevents unchecked collagen secretion and fibrosis by preventing myofibroblast proliferation and earmarking them for subsequent immune clearance…Moving forward in our myofibroblast panel, we observed that expression of TGF-β cytokine, TGF-β1, decreased significantly with replicative senescence in both RNA and protein. However, we see robust induction of the TGF-β isotype 2 (TGF-β2) cytokine with replicative senescence as TGF-β1 abundance drops, which indicates a switch in TGF-β isotypes with replicative senescence. It has been shown TGF-β2 is a more potent inducer of the endothelial to mesenchymal transition (EndMT) in vitro compared to TGF-β1 and TGF-β3 in human microvascular endothelial cells, and TGF-β2 may be playing a similar role here in inducing FMT and replicative senescence in WI-38 cells.
It occurs to me that the beneficial action of TGF-B activating GDF11 may be converting transdifferentiated myofibroblasts back to the cell they should be, by rebalancing TGF-B isotypes. They also found a drug that blocks much of the TGF-B replicative senescence signalling through YAP1/TEAD1 (although I doubt this would be a plausible treatment for aging, see VERTEPORFIN).
Furthermore, we found that a subset of YAP1/TEAD1 targets are both induced with replicative senescence, and are principal components of TGF-β signaling. Thus, the YAP1/TEAD1 complex may be acting in convergence with TGF-β signaling with increasing PDL to enact a myofibroblast-like state that we recognize as replicative senescence…We found that treatment with 10 µM verteporfin for 2 hours reduced expression of these three YAP target genes by 70% relative to the no treatment control.
Some final choice quotes from the discussion section.
Importantly, very little changed across all modalities in the hTERT cell line.
No cellular aging if telomeres don't shorten.
…we provide evidence that the early manifestation of the senescent gene expression reflects gradual changes on a per cell basis rather than changing cell proportions. In effect, individual cells ‘show their age' with increasing PDL long before permanently exiting the cell cycle and transiting fully into the senescent state. The implications of this conclusion extend to organismal aging.
The importance of this paragraph for understanding aging cannot be overstated. In other words, it's no good just looking for specific markers for senescent cells. By inference senolytics will also not be a panacea.
Importantly, the gradual progression suggests that cells need not reach the endpoint to elicit a phenotype. For instance, proliferative fibroblasts isolated from IPF patients exhibited multiple senescent features and phenotypes in addition to accelerated senescence progression.
Perhaps measures of methylation aging are capturing some of this phenomenon, long before cellular senescence is reached.
Lastly, this phenomenon is not constrained to fibroblasts as we observe that the salient regulatory features of replicative senescence extend to cell types as distant as astrocytes.
It's not just fibroblasts. It is likely all cycling cells that suffer telomere loss.
We hypothesize that during fibrotic disease states and/or age, fibroblasts migrate to sites of micro-injuries. As these proliferating fibroblasts become replicatively aged, they are triggered (by DNA damage or other insults) to rewire their metabolism to induce FMT via active epigenetic reorganization.
In other words, aging is state whereby fibroblasts are trying to repair the body all over, leading to widespread conversion into myofibroblasts.
It is important to note here that the in vitro DNA damage here arises primarily from telomere erosion documented by the observation that our hTERT control cultures do not exhibit the same changes.
And there you have it. Despite barely mentioning telomeres until this point, this team have gone much of the way to proving the telomere therapy of aging. Thank you (Calico) and good night.
Edited by QuestforLife, 05 April 2022 - 03:55 PM.