































Researchers are hijacking the cell’s protein-disposal system in the fight against Alzheimer’s and intractable cancers.

When Craig Crews first managed to make proteins disappear on command with a bizarre new compound, the biochemist says that he considered it a “parlour trick”, a “cute chemical curiosity”.
Today, that cute trick is driving billions of US dollars in investment from pharmaceutical companies such as Roche, Pfizer, Merck, Novartis and GlaxoSmithKline. “I think you can infer that pretty much every company has programmes in this area,” says Raymond Deshaies, senior vice-president of global research at Amgen in Thousand Oaks, California, and one of Crews’s early collaborators.
The drug strategy, called targeted protein degradation, capitalizes on the cell’s natural system for clearing unwanted or damaged proteins. These protein degraders take many forms, but the type that is heading for clinical trials this year is one that Crews, based at Yale University in New Haven, Connecticut, has spent more than 20 years developing: proteolysis-targeting chimaeras, or PROTACs.
Large and unwieldy, PROTACs defy conventional wisdom on what a drug should be. But they also raise the possibility of tackling some of the most indomitable diseases around. Because they destroy rather than inhibit proteins, and can bind to them where other drugs can’t, protein degraders could conceivably be used to go after targets that drug developers have long considered ‘undruggable’: cancer-fuelling villains such as the protein MYC, or the tau protein that tangles up in Alzheimer’s disease.
“This is new territory,” says Alessio Ciulli, a biochemist at the University of Dundee, UK. “We’re breaking the rules of what we thought would be druggable.”
The field has reason to be optimistic. In 2014, scientists discovered that the myeloma treatment lenalidomide (Revlimid), one of the world’s best-selling drugs, works in a similar way to protein degraders to chew up two formerly untouchable proteins1,2.
Yet the field lacks published data confirming that PROTACs and other emerging compounds can make undruggable proteins disappear. And there are questions about where and how these odd-looking molecules will work in the body.
For now, all eyes are on Arvinas, a biotech company in New Haven, Connecticut, founded by Crews. It’s scheduled to begin testing a PROTAC for prostate cancer, albeit attacking a protein that’s been targeted successfully by other drugs. “We’re on the cusp of proving these PROTACs can be drugs,” says Ian Taylor, senior vice-president of biology at Arvinas. “Right behind that will be: can we do this with an undruggable?”.
An academic exercise
In diagrams, PROTACs often look like dumb-bells. They are molecules made up of two binding ends connected by a thin tether.
The action happens on the ends. One grabs on to the target protein, while the other latches on to a ubiquitin ligase — part of the cell’s natural rubbish-disposal system that labels defective or damaged proteins by slapping a small protein called ubiquitin onto them (see ‘Marked for destruction’). Ubiquitin tags act as sort of ‘Please collect’ stickers that instruct the cell’s protein shredder, called the proteasome, to do its thing.
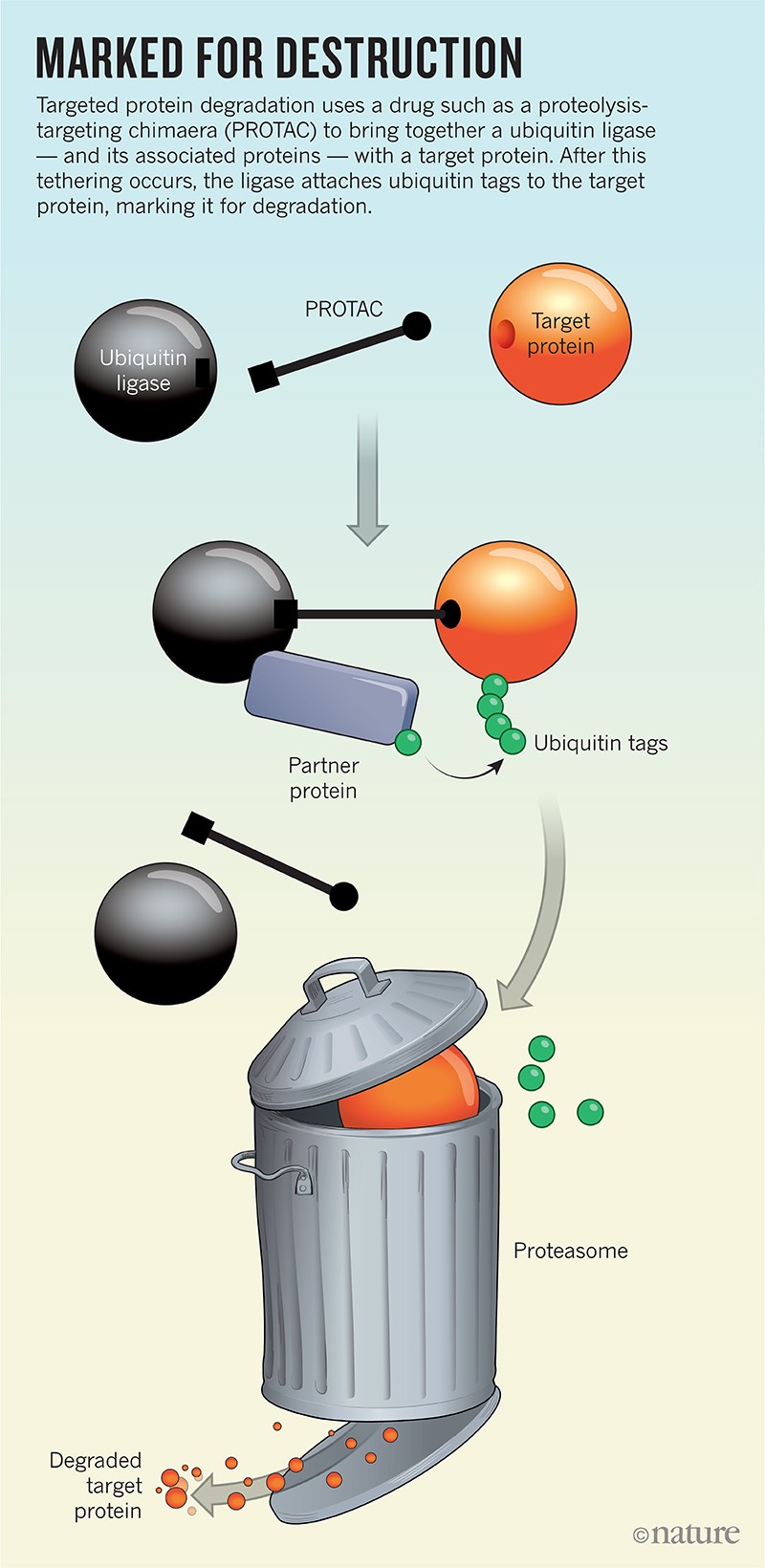
Proximity accounts for a lot in biology, so by simply bringing together the ligase and the target protein, a PROTAC ensures that the target will get marked for destruction. Ligases are efficient and ubiquitin, as the name suggests, is plentiful, so a single PROTAC should be able to perform its catch-and-release function repeatedly throughout the cell, suggesting that only a small amount of such a drug is needed for potent activity.
The earliest-known published description of a PROTAC-like molecule is in a patent filed in 1999 by two scientists at Proteinix, a biotechnology company in Gaithersburg, Maryland. In the patent (see go.nature.com/2vyjf9l), John Kenten and Steven Roberts proposed co-opting the cell’s protein-degradation system. Colleagues dismissed the idea, saying that Kenten and Roberts were complicating drug discovery by trying to bind to two proteins — the unwanted protein and the ligase — at once. “There was not a lot of enthusiasm internally for it,” recalls Kenten, now research director at Meso Scale Diagnostics in Rockville, Maryland. Proteinix did not pursue the approach.
But on the other side of the United States, another pair of minds was mulling the same idea. During a research retreat in 1998 at a scenic resort on Semiahmoo Bay in northwest Washington, Deshaies paused in front of a poster by Crews to listen to him talk about using small molecules to link two proteins together. Deshaies, then a biochemist at the California Institute of Technology in Pasadena, was knee-deep in the study of ubiquitin ligases. The human genome encodes roughly 600 of them, which need to form a complex with other proteins to do the tagging. About a year earlier, Deshaies had co-discovered3 a protein family now known to contain 250 ubiquitin ligases.
“It wasn’t that big of a leap to come to the idea of, well, gee, if you could link things to ubiquitin ligases then you could potentially drive the ubiquitination of a protein — and its degradation,” recalls Deshaies. He and Crews continued to chat all weekend and parted ways with a plan to find funding to explore the idea.
At the time, Crews was developing a drug that worked in the opposite way to PROTACs. It blocked the ubiquitin system in cells, causing proteins to build up to dangerous levels and eventually trigger cell death. The result of that work, carfilzomib (Kyprolis), is now used to treat the blood cancer multiple myeloma. “I thought the flip side would be equally as interesting,” says Crews. “That certainly has turned out to be the case.”
Crews and Deshaies soon published a study demonstrating that their first PROTAC, Protac-1, successfully grabbed and led to the degradation of a cancer-associated protein called METAP2 in extracts from Xenopusfrog eggs4.
Still, Protac-1 was far from being a drug, says Crews, who called the paper an “academic exercise”. First-generation PROTACs had low activity in human cells, probably because the compounds struggled to get inside. They relied on big, unwieldly peptides to bind to the ligases. The scientists had to find a way to make the ligase-binding ends more drug-like — “Something that had potential to be a pharmaceutical,” says Crews. Or they needed to move on.
With funding and research support from GlaxoSmithKline in London, Crews pushed ahead, mainly targeting one particular ligase, the von Hippel–Lindau disease tumour suppressor (VHL). In 2012, Crews, together with his graduate student Dennis Buckley and Ciulli, a former visiting fellow in Crews’ lab, reported on a small-molecule binder for VHL5. Crews finally began to believe that PROTACs really could become drugs.
Fishing for small molecules
Crews wasn’t the only one chasing protein degraders. In 2010, while at the Dana-Farber Cancer Institute in Boston, Massachusetts, chemical biologist James Bradner read a paper by a team of researchers in Japan, led by Hiroshi Handa, then at the Tokyo Institute of Technology in Yokohama6. Handa had been trying to understand why the infamous drug thalidomide, approved in some countries in the late 1950s and early 1960s to help with nausea in pregnancy, caused problems with limb development. (It is now approved to treat multiple myeloma and a skin condition.) Using thalidomide as the bait to fish for proteins in cells, Handa discovered that the drug hooks on to and blocks the activity of a ubiquitin ligase called cereblon. That inhibition, his team found, affects limb growth and development in zebrafish and chicks6.
Bradner realized that if thalidomide binds to a ubiquitin ligase — no easy feat, because such enzymes are notoriously difficult to grab — then perhaps he could find a way to bind to the same ligase but target it to proteins implicated in disease. In 2013, Buckley joined Bradner’s team as a postdoctoral researcher, and they began the search for small molecules that bind to cereblon.
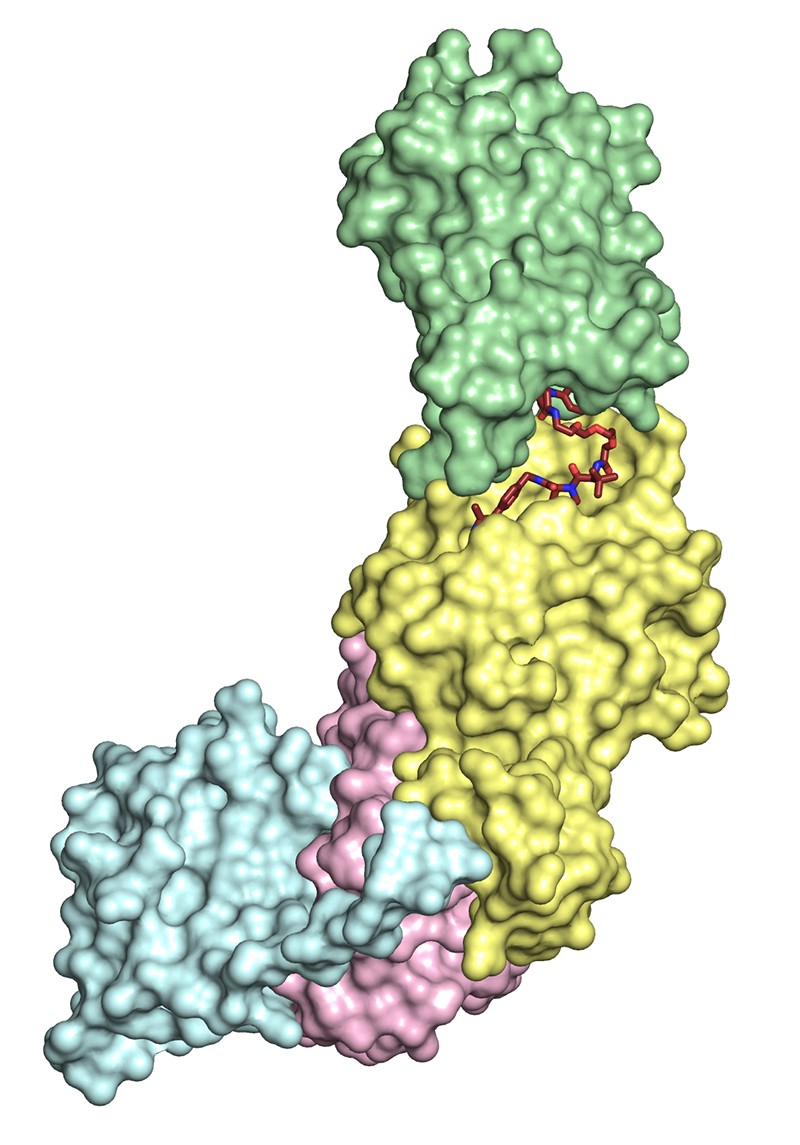
The PROTAC MZ1 bound to a ubiquitin-ligase complex (yellow, pink and blue) and its target protein BRD4 (green).
In May and June 2015, three teams — led by Bradner, Ciulli and Crews — published five separate papers describing small-molecule PROTACs with potent, drug-like activity7–11. With Ian Churcher at GlaxoSmithKline, Crews bound a PROTAC to VHL and used it to degrade the levels of several proteins to less than 10% of those present in untreated cells7. Bradner and his colleagues bound cereblon to their PROTAC to reduce levels of a cancer-causing protein8, and Ciulli, by then at the University of Dundee, and his team degraded the same protein, using VHL as the ligase9. The protein degraders worked both in cells in a dish and in human tumours in mice.
As well as designing drug-like protein degraders, Crews and Bradner’s teams have both built systems — HaloPROTACs10 and dTAG12, respectively — that enable researchers to put targeted protein degradation to work as a tool in the laboratory, using genetic tags to mark proteins for destruction in cultured cells and in mice. With dTAG, “you can deplete a protein in minutes or hours and monitor what happens”, says Behnam Nabet, a chemical biologist who led development of the system with Nathanael Gray at the Dana-Farber Cancer Institute. “This gives you a lot of power to study oncogenes and kinases and proteins that have very rapid activity.” The dTAG materials are currently freely available: more than 150 academic labs use the probe to investigate the effects of depleting specific proteins in cells, says Nabet.
Bradner, who left Dana-Farber in 2016 to become president of the Novartis Institutes for Biomedical Research, estimates that around 30 separate tools already incorporate the technology. “The path to chemical probes is now well established,” he says. “But the challenge to make real-world medicines from these ligands is significant.”
Gold rush
Following the 2015 flurry of small-molecule PROTACs, Deshaies, who had left the field, penned an opinion piece declaring that PROTACs had the potential to become a major new class of drug, possibly surpassing two of the hottest drug-development areas of all time — protein kinase inhibitors and monoclonal antibodies13. “The gold rush is on!” Deshaies wrote at the time.
Since then, he says, it has only intensified. He joined Amgen in 2017 and now oversees the company’s work in the area.
The Arvinas trial, expected to begin by mid-2019, will include 28–36 men with metastatic prostate cancer and will last around 9 months, says Taylor. It is usual for any new class of drug to go after a well-known target, where the biology and toxicology are well-understood, and Arvinas’s first candidate is no exception. It degrades the androgen receptor, a protein that is already targeted by a handful of approved drugs. The company hopes that by degrading rather than inhibiting the receptor, its PROTAC will be able to treat people who have become resistant to or see no benefit from existing drugs. And if the candidate succeeds, the field will finally have the clinical data that everyone is looking for. Arvinas will have shown that a PROTAC can be a drug.
That’s crucial because there has been considerable doubt about whether protein degraders can work in humans. Fully assembled PROTACs break well-known rules of thumb for drugs. Chief among them is size. A good small-molecule drug typically has a mass of less than 500 daltons. Current PROTACs range upwards of 1,000 daltons. Yet the molecules can still enter cells7,10,11. Crews suspects that this is because they are probably recognized by the cell membrane as two smaller molecules that happen to be tethered together, rather than a single large one.
“We’re throwing out preconceived notions we’ve had about larger-than-average small molecules,” says Taylor.
Also out of the window are preconceived ideas about undruggables. The problem with many of these tough protein targets is that most small-molecule drugs or monoclonal antibodies need to bind to an active site on an enzyme or a receptor to work. But an estimated 80% of proteins in human cells lack such a site. PROTACs, however, can grab a protein by any nook, cranny or crevice — they don’t need to be sitting in an active pocket to work. So they could make those proteins accessible.
There’s already some evidence to support this approach. Last year, a team at the Institute of Cancer Research in London produced a small molecule that can bind to a transcription-factor regulator that doesn’t have an active site14. They were able to create a potent PROTAC by attaching a binder for the ubiquitin ligase cereblon.
The field still lacks published evidence of a PROTAC that can target and degrade a valuable undruggable protein. Deshaies says that Amgen has a PROTAC effective in both cultured cells and animals against an unnamed high-value cancer target that has been historically tough to bind. Arvinas claims to have in vivo evidence of PROTACs degrading tau in the brains of mice. On its website, the company says that injecting its tau-protein degrader directly into the mouse hippocampus reduced levels of tau by 50%.
By developing PROTACs for an array of diseases, including those that affect the brain, Taylor says that many researchers hope to show that the technology is “therapeutic-area agnostic”. Various teams are also working to expand the pool of ligases that protein degraders can recruit. There are only four main ones used at present, including VHL and cereblon, and a wider variety of available ligases could enable drug developers to match the most potent ligase–PROTAC combination with their cell type or protein of interest. “Potentially, any ligase can be hijacked through this approach,” says Ciulli, who is collaborating with German pharmaceutical company Boehringer Ingelheim on the development of PROTACs.
Buoyed by fresh targets, improved potency, and a clinical trial about to begin, researchers are ready to prove that protein degraders can be more than a parlour trick. “The sky is the limit,” says Ciulli. “It is just a question of when.
Source: https://www.nature.c...586-019-00879-3

















