You may recall from a distant high-school science class that mitochondria are the “powerhouse of the cell.” This organelle is essential because it produces energy for cells, so you might think the more mitochondria, the better—but not if they’re damaged. New research published in Nature Neuroscience shows that damaged mitochondria may play a critical role in neurodegenerative disorders like Alzheimer’s disease, suggesting new avenues for possible treatments.
.jpg?auto=compress%2Cformat)
Two mitochondria, the “powerhouse of the cell” and potential therapeutic target for neurodegenerative diseases
When healthy, mitochondria are little engines, producing a molecule called ATP that’s like a battery for the cells’ energy needs. This is especially important in the brain, which uses up to 20 percent of the body’s energy. But when mitochondria are damaged, they wreak havoc by producing large quantities of reactive oxygen species, by-products of oxygen breakdown that are toxic to neurons. This kind of mitochondria dysfunction is one of the processes observed in Alzheimer’s disease and other neurodegenerative diseases.
Normally, cells have a process for getting rid of faulty mitochondria, called mitophagy. During mitophagy, the cell recognizes and removes damaged mitochondria. Without mitophagy, you get an accumulation of defective mitochondria, causing neuron injury and death. Adding insult to injury, mitophagy is hampered in Alzheimer’s disease.
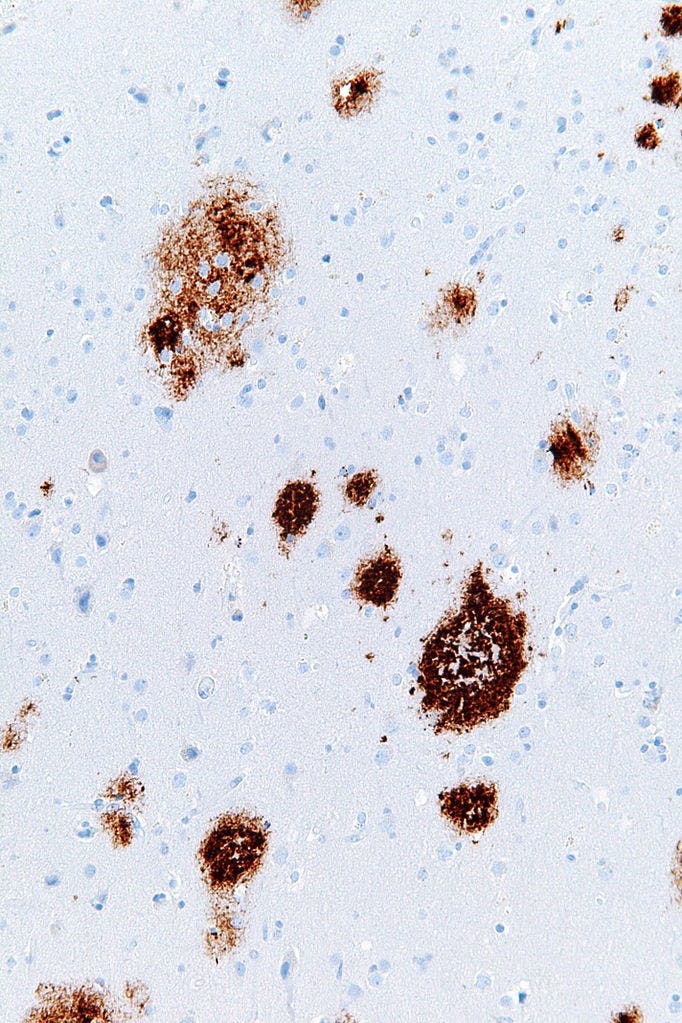
Plaques (brown) in tissue from a patient with Alzheimer’s disease.
Scientists have long speculated that Alzheimer’s disease is driven by clumps of a protein called amyloid-beta plaques. This hypothesis suggests that these plaques build up, leading to the accumulation of another protein called tau. Together these protein collections eventually kill neurons and impair memory. Studies now suggest why that happens—it turns out mitochondrial dysfunction can activate processes that cause these dangerous stockpiles of amyloid-beta and tau.
This research supports the mitochondrial cascade hypothesis, proposed in 2004 by Russell Swerdlow and Shaharyar Khan. Swerdlow and Khan were the first to suggest that Alzheimer’s disease actually starts with the mitochondria, not with amyloid-beta. In other words, mitochondria dysfunction may be the root problem— which would fundamentally change the way the we’re searching for Alzheimer treatments. Indeed, impaired mitochondrial function precedes amyloid-beta accumulation in mouse models of disease. If this hypothesis proves true, destroying defective mitochondria could be a way of curing the disease.
Armed with this new insight, Vilhelm Bohr at the National Institute on Aging led an investigation to see if activating mitophagy and clearing out damaged mitochondria helped improve learning and memory. First, the researchers evaluated a panel of compounds to find what could encourage mitophagy in the worm C. elegans. They found two compounds, actinonin and urolithin A, that effectively activated this process. By inserting the genes that trigger expression of amyloid-beta and tau, researchers are able to mimic Alzheimer’s disease in worms. Treating these worms with the compounds successfully removed their damaged mitochondria—and reduced levels of amyloid-beta and phosphorylated tau, a version of tau which is more prone to aggregation.
As a first step, this was encouraging: Simply by activating mitophagy, they got rid of the features of Alzheimer’s disease in worms.
But although worms are pretty cool to study, they’re fairly different than humans. Translating results to vertebrate animals is crucial, especially if there is the possibility of identifying a new potential treatment. So next, researchers tried a similar approach on mouse models. These rodents are engineered to have the mutations present in human Alzheimer’s disease patients and have the hallmark memory loss associated with the disease. Like the worms, in the mice models, mitophagy activators decreased the number of defective mitochondria in the mice’s brains, as well as their dangerous protein accumulation.
Mitophagy activation reduced these toxic proteins using a surprising type of cell—microglia. These are the immune cells of the central nervous system—they get rid of debris and other unwanted components in the brain, and have a particularly important role in Alzheimer’s disease, where they have been shown to engulf or “eat up” the amyloid-beta plaques.
Bohr’s team found that mitophagy activators helped microglia better destroy existing amyloid-beta plaques in the mice’s brains. Basically, inducing mitophagy made the microglia better “eaters.” Microglia have been studied extensively in the field, and this study only underlines their importance in Alzheimer’s disease. It seems that mitochondrial health may be critical in how well microglia are able to engulf amyloid-beta plaques.
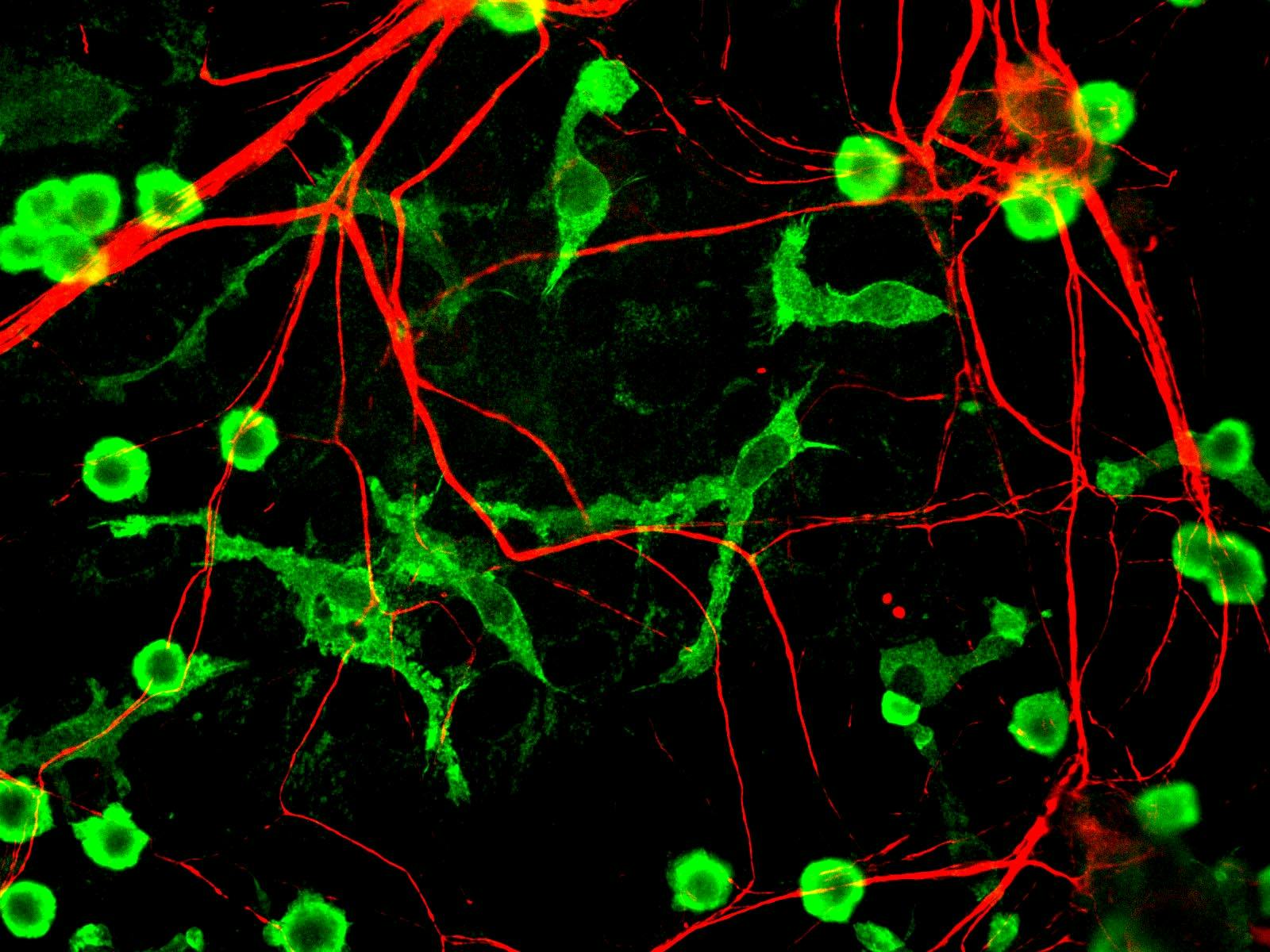
Microglia (in green) associate closely with neurons (in red).
This is all very promising, but the main symptom of Alzheimer’s disease is memory impairment. The scientists therefore needed to see if it also had an effect on the learning processes of the animals. Fortunately, it turns out that worms treated with mitophagy activators remembered better than worms that had not been treated with the activators. Similarly, mitophagy activator-treated mice were better able to remember the context associated with a stimulus in different learning tests. This suggests the treatment improves learning and reduces memory impairment.
But these studies have not yet confirmed if mitophagy was activated only in the brain, or if it was also induced in other organs. Although activators appear to have beneficial effects on the brain, further research will be needed to discover if increasing mitophagy might negatively affect other systems elsewhere in the body. For example, could these compounds also get rid of “healthy” mitochondria? If over-activation of mitophagy gets rid of too many mitochondria, it could lead to energy deficiency and other health impacts.
Another unanswered question is how mitochondria and mitophagy become defective in the first place. Research shows that amyloid-beta and tau accumulations can lead to mitochondria dysfunction, but it’s still a chicken-or-the-egg question: Which comes first? There may be a vicious cycle occurring between mitophagy and protein aggregation, where amyloid-beta plaques and tau tangles can worsen mitochondrial dysfunction, and vice versa.
We now have powerful evidence suggesting that accumulation of faulty mitochondria contributes to events that lead to Alzheimer’s disease. By eliminating the damaged organelles through mitophagy, you also alleviate the symptoms related to the disease. Further research is needed, but the implications are huge: It suggests a new therapeutic target for people with Alzheimer’s disease. The two mitophagy inducers, actinonin and urolithin A, are an antibiotic and a compound that derives from the breakdown of molecules found in pomegranates, respectively. So it may be relatively quick to get them approved for clinical trials in humans.
This study suggests that Alzheimer’s disease may actually begin with compromised mitochondrial function—a dramatic change in how the disease has been understood. This may be the reason why drugs that target the production of amyloid-beta plaques have been disappointing in clinical trials—it’s because they are not the primary problem. With more studies like these, we may begin thinking differently about how we look at and treat Alzheimer’s disease, offering hope for new treatments.
Source: https://massivesci.c...plaques-memory/