































.
T I M E L I M I T E D A C C E S S S O U R C E : nature
On average, an approved drug today costs $2-3 billion and takes over ten years to develop. In part, this is due to expensive and time-consuming wet-lab experiments, poor initial hit compounds, and the high attrition rates in the (pre-)clinical phases. Structure-based virtual screening (SBVS) has the potential to mitigate these problems. With SBVS, the quality of the hits improves with the number of compounds screened. However, despite the fact that large compound databases exist, the ability to carry out large-scale SBVSs on computer clusters in an accessible, efficient, and flexible manner has remained elusive. Here we designed VirtualFlow, a highly automated and versatile open-source platform with perfect scaling behaviour that is able to prepare and efficiently screen ultra-large ligand libraries of compounds. VirtualFlow is able to use a variety of the most powerful docking programs. Using VirtualFlow, we have prepared the largest and freely available ready-to-dock ligand library available, with over 1.4 billion commercially available molecules. To demonstrate the power of VirtualFlow, we screened over 1 billion compounds and discovered a small molecule inhibitor (iKeap1) that engages KEAP1 with nanomolar affinity (Kd = 114 nM) and disrupts the interaction between KEAP1 and the transcription factor NRF2. We also identified a set of structurally diverse molecules that bind to KEAP1 with submicromolar affinity. This illustrates the potential of VirtualFlow to access vast regions of the chemical space and identify binders with high affinity for target proteins.
Repeated optimization of lead compounds and late-stage failure of drug candidates are the primary causes of longer development times and increased costs in drug development. Improving the quality of the initial lead compounds would minimize these lead optimization cycles and result in drug candidates entering (pre-)clinical phases with greater specificity and higher affinity. Virtual screening to identify molecules that bind to a specified site on a receptor protein has become an important part of the drug discovery pipeline2–5. Current virtual screening paradigms routinely sample only a tiny fraction, on the order of 106-107 molecules, of the total chemical space of small organic compounds suitable for drug discovery, estimated to encompass more than 1060 molecules6. However, the scale of a virtual screen is of central importance because the more compounds that are screened, (a) the lower the rate of false positives, and (b) the more favourable the quality of the lead compounds (e.g. higher-affinity binders). It was recently shown experimentally that ultra-large scale screening improves the rate of true positives2. Here we derived a probabilistic model of the true-positive rate as a function of the number of compounds screened, and analysis of our ultra-large screen confirms that the docking score of the highestscoring compounds improve with the scale. Increasing the scale of a virtual screen can improve the quality of initial hits in two distinct ways: (1) by identifying hits with tighter binding affinity, which can result in lowered dosages and fewer off-target effects, and (2) by discovering compounds with more favourable pharmacokinetic and/or less inherent cytotoxic properties.
To increase the number of compounds evaluated in a virtual screen by orders of magnitude and make it accessible to any researcher, there is a dire need for a platform that can integrate all the tasks in the virtual screening process. Such a platform should ideally (1) scale linearly with the number of CPUs, (2) efficiently handle billions of files, (3) minimize input and output (I/O) load, (4) run robustly (e.g. skip incorrectly encoded ligands, resist temporary I/O problems, and resume following unexpected termination), (5) run on any type of computing cluster (including cloud platforms), and (6) be user-friendly and easy to use for non-computational scientists. Furthermore, to provide flexibility, a SBVS platform should be able to interface with a variety of docking programs, support both rigid and flexible receptor docking, test multiple docking scenarios in a single workflow, allow for consen-sus and ensemble docking, and carry out multiple replicas of the same docking scenario. Lastly, to democratize access, facilitate widespread usage, and catalyse further development, such a platform would need to be open source.
With these requirements in mind, we designed VirtualFlow, an open-source platform that is able to screen chemical space on an unprec-edented scale. Screening one billion compounds on a single processor core, with an average docking time of 15 seconds per ligand, would take 475 years. By contrast, VirtualFlow can dock one billion compounds in approximately two weeks by leveraging 10,000 CPU cores simultane-ously. Such high performance computing facilities are available to researchers through several potential sources, including local insti-tute computer clusters, national super-computing centres, or cloud computing platforms.
Targeting KEAP1 using VirtualFlow
To test the advantages of ultra-large-scale in silico screening and the performance of the VirtualFlow platform we decided to target the chal-lenging and therapeutically relevant protein-protein interaction (PPI) between nuclear factor erythroid-derived 2-related factor 2 (NRF2) and Kelch-like ECH-associated protein 1 (KEAP1). NRF2 is a master regula-tor of cellular resistance to oxidative stress and cellular repair7. Under unstressed conditions, NRF2 is sequestered by KEAP1, an E3 ubiquitin ligase substrate adaptor, and targeted for degradation8. However, upon oxidative stress, reactive oxidants dissociate NRF2 from KEAP1 and NRF2 translocates to the nucleus to activate its transcriptional program of approximately 250 genes9. The NRF2-KEAP1 pathway is critical in protecting the cell under oxidative stress and inflammation and is implicated in a number of diseases10. There are ten drugs target-ing KEAP1 that are in clinical trials and nine more that are at the pre-clinical stage10. Using VirtualFlow, we screened ~1.3 billion compounds (~1 billion compounds from the Enamine REAL Library and ~330 mil-lion compounds from the ZINC library) against the NRF2 interaction interface on KEAP1. First, we would like to describe the salient features of VirtualFlow and its scalability.
Characteristic features of VirtualFlow
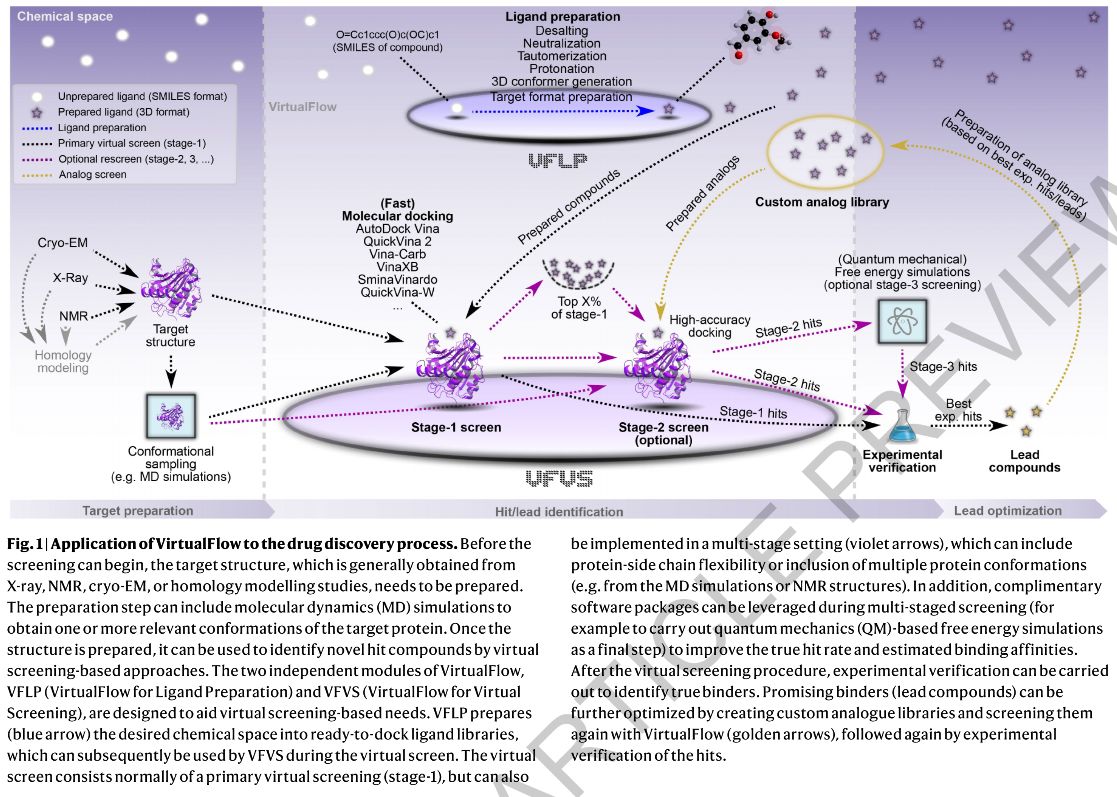
One of the key features of VirtualFlow is its linear scaling behaviour (O(N)) with respect to the number of CPUs and nodes utilized. Virtu-alFlow can run on computer clusters operated with any of the major resource managers (SLURM11, Moab/TORQUE12, PBS13, LSF14 and SGE15), and compatibility with additional job schedulers can be easily added. Thus VirtualFlow is also ideally configured for cloud computing plat-forms like Amazon’s Web Services (AWS), Microsoft’s Azure and Goog-le’s Cloud Platform (GCP). VirtualFlow is able to run autonomously from the first to the last ligand in the screening pipeline, a feature facilitated by automatic submission of new batch system jobs. The workflow can be monitored and controlled in real time. The VirtualFlow package consists of two applications that work seamlessly together: The VFLP (VirtuaFlow for Ligand Preparation) module, which prepares small molecules for screening; and the VFVS (VirtualFlow for Virtual Screening) module, which executes the virtual screening procedures (Fig.1). The separation of ligand preparation and virtual screening is desirable because the same ready-to-dock ligand library can be used in any number of VFVS virtual screens.VirtualFlow for Ligand Preparation (VFLP)VFLP (VirtualFlow for Ligand Preparation) prepares ligand databases by converting them from the SMILES format into any desired target format (e.g. the PDBQT format, which is required by many of the AutoDock-based docking programs). VFLP uses ChemAxon's JChem package as well as Open Babel to desalt ligands, neutralize them, generate (even multiple) tautomeric states, compute protonation states at specific pH values, calculate 3D coordinates, and convert the molecules into desired target formats (Extended Data Fig.2). The output file formats currently supported by VFLP are shown in Supplementary Table7.
Preparation of the Enamine REAL library
Commercially available compounds constitute the most interesting subset of the chemical space, since these compounds can be readily purchased. The largest vendor library available today is the REAL library of Enamine, containing approximately 1.4 billion make-on-demand compounds (as of October 2019 the ZINC 15 database contained 1.46 billion compounds, but only provided 630 million molecules in a ready-to-dock format). We have used VFLP to convert the ~1.4 billion compounds of the REAL library into PDBQT format (seeMethods), and have made it freely available on the VirtualFlow homepage, accessible via a graphical interface (Supplementary Fig.5). The entire database has a six-dimensional lattice architecture, the general concept of which was modelled after the ZINC 15 database16, where each dimension cor-responds to a physico-chemical property of the compounds (molecular weight, partition coefficient, number of hydrogen bond donors and acceptors, number of rotatable bonds, and the topological polar sur-face area). The preparation of ligands using VFLP is a one-time effort.
VirtualFlow for Virtual Screening (VFVS)
To set up a virtual screen with VFVS, a set of docking scenarios is speci-fied by the user. Docking scenarios are defined by the choice of the external docking program, the receptor structure, and the docking parameters (which include the pre-defined docking surface on the receptor, residues on the receptor that are allowed to be flexible during docking, and the rigor of the docking routine). VirtualFlow currently supports the following docking programs: AutoDock Vina17, QuickVina 2[18], Smina (which includes the Vinardo and AutoDock 4 scoring func-tions)19, AutoDockFR (ADFR)20, QuickVina-W5, VinaXB21, and VinaCarb22. By supporting an array of different docking programs, VFVS can be used in a variety of cases by leveraging the unique advantages of each program. VFVS allows the specification of multiple docking scenarios to be carried out for each ligand, enabling consensus docking proce-dures, as well as ensemble docking procedures23,24. VirtualFlow is also amenable to the integration of other docking programs that are not currently a part of this platform.
Scaling behaviour of VFVS
In order to measure the scaling behaviour of VFVS, we measured the performance on two local clusters, LC1 and LC2. On LC1, we used 18,000 CPU cores of heterogeneous composition (different models of Intel Xeon and AMD Opteron processors), whereas on LC2 we employed up to 30,000 Intel Xeon 8268 CPUs. The scaling behaviour was effectively linear in both cases (i.e., O(N), where N is the number of cores), see Extended Data Fig.3a. These results meet theoretical expectations since there is no direct communication between the processes running in parallel, which is key to perfect scaling behaviour without bounds. The independence of its parallel processes means that VirtualFlow is expected to scale linearly even if millions of cores are used. We also tested the performance of the platform on cloud-based computing systems including GCP and AWS. On the GCP we carried out large-scale benchmarks with up to 160,000 CPUs, and despite this massive scal-ing in CPU volume, VirtualFlow still exhibited linear scaling behaviour (Extended Data Fig.3a). A typical high-throughput screen, such as the one described in this study, of 1 billion compounds will take ~15 hours on the GCP with 160,000 CPUs, making VirtualFlow suitable for the highly anticipated exascale computing age.
Multistaged virtual screens with VFVS
VFVS can also be used to organize virtual screens with multiple stages to substantially increase the quality of the results (Fig.2a). In the multi-staging approach, several virtual screens are executed in succession. The number of top-scoring compounds that advance from one stage to the next is successively reduced, with concomitant increases in docking accuracy and computational cost.
Using VFVS to screen 1.3 billion ligands
In order to validate the performance of VFVS we screened a virtual library of 1.3 billion commercially available compounds (~330 million compounds from the ZINC 15 database16, and ~1 billion compounds from the Enamine REAL library) against KEAP1. %It should be noted that there is some overlap of compounds between the two libraries.This effort was completed in around four weeks, using on average approximately 8,000 cores on a heterogeneous Linux cluster.To illustrate the benefit of an ultra-large-scale screen, we chose a subsets of the ligands (0.1, 1, 10, and 100 million compounds) randomly from the ~1 billion compound screen of the REAL library and consid-ered the scores of the top 50 compounds.(Fig.2b). As the scale of the screen increased, the average docking score increased thus improving the chances of identifying tighter binders. This in turn leads to higher true hit rates and tighter experimental binding affinities, as predicted by a probabilistic model which we derived (Supplementary SectionD), and which experimentally demonstrated previously2.To demonstrate VirtualFlow in a multi-staging context we subjected the top ~3 million ranking compounds from the primary virtual screen to a rescoring procedure (Fig.2a). In stage-2, the 13 residues of KEAP1 at the NFR2 interaction interface were allowed to be flexible. This flex-ibility accounted for the movement/dynamics of the amino acids at the binding interface, not captured by a static structure. In the re-scoring procedure we utilized two different docking programs (Smina Vinardo and AutoDock Vina), and two replicas of each docking scenario were carried out to further increase the conformational space sampled dur-ing the docking runs. The necessity of multi-stage screening depends on the target of choice and the computational resources available, but this type of virtual screen is particularly useful in cases where dynamics at the docking interface is expected to play a significant role.
Experimental validation
From the in silico screen described above, we chose 590 hits for experi-mental validation. Of these, 492 compounds were from the top 0.03% of stage-2 screen and 98 compounds were from the top 0.0001% of stage-1. Hits from stage-1 were ordered to compare the true hit rate between stage-1 and stage-2 hits, in a multistage setting. In addition to the ranking by docking score, the choice of these compounds was based on factors like drug-likeness, availability for procurement, ligand efficiency and chemical diversity. We used four established biophysical methods: fluorescence polarization (FP), surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), and bio-layer interferometry (BLI) to experimentally validate the binding of the VirtualFlow-derived hits to KEAP1. FP and SPR were initially used in a high-throughput fash-ion (Level-1) to detect binding and the compounds identified here were subsequently validated with more scrutiny in a detailed and low-throughput fashion (Level-2). We used a recombinantly expressed and purified Kelch domain of mouse KEAP1, henceforth referred to as KEAP1. An overview of the experimental verification workflow is graphi-cally represented in Extended Data Fig.6, and a detailed description of the experimental procedure is provided in the methods section. Of these four biophysical methods, FP and BLI detect the ability of the hits to displace the NRF2 peptide from KEAP1, identifying hits we refer to as displacers. SPR and NMR directly detect binding of hits to KEAP1, identifying hits referred to as binders. VirtualFlow identifies molecules that potentially bind to the NRF2-interacting interface on KEAP1, but the in silico screen is performed using KEAP1 alone, in the absence of NRF2. The NRF2 binding surface on KEAP1 is part of the deep pocket/tunnel of the KEAP1 β-barrel with NRF2 binding to the entrance of this tunnel. However, some compounds could bind more tightly by inserting deep into this central tunnel of KEAP1 rather than embracing the surface like the NRF2 peptide, and/or bind to parts of KEAP1 not engaged by NRF2. Such binders might not effectively disrupt the interaction with NRF2, while still engaging KEAP1 with high affinity (Extended Data Fig.9). In our experimental validation we identified both displacers and binders.Out of the cherry-picked 590 compounds, 69 were confirmed to bind to KEAP1 by Level-2 SPR. To assess the ability of the compounds to displace the NRF2 peptide we used the FP assay. Ten compounds were confirmed to be displacers with an IC50 < 60 µM by FP and all of them were identified as a binder by Level-2 SPR. Interference by auto-fluorescence from the compounds themselves prevented the analysis of some of the compounds by FP. Thus, we used BLI as an orthogonal assay to assess the ability of the compounds to displace NRF2. The binding affinity of the NRF2 peptide to KEAP1 as measured by BLI was 1.86 nM which is similar to that measured by FP, 3.67 nM (Extended Data Fig.4). 40 compounds of the 69 SPR Level-2 active compounds were able to disrupt the NRF2-KEAP1 interaction as observed by BLI. Of these 40 compounds, 16 were able to displace NRF2 from KEAP1 at a compound concentration of 20 µM, while all 40 compounds could do so at 100 µM. Using BLI, we were able to identify displacers that were missed by FP due to autofluorescence (an example is shown in Extended Data Fig.8). We tested all the SPR Level-2 active compounds for potential aggregation by Dynamic Light Scattering (DLS). We identi-fied seven compounds that aggregated in the DLS assay and hence were not considered for further evaluation (Supplementary Table5). Based on the SPR Level-2 and the FP Level-2 binding data, we selected 23 com-pounds for SPR Level-3 experiments to determine the binding affinity. All 23 compounds had affinities in the low micromolar to nanomolar range, and 12 compounds had submicromolar Kd values. From these 23 compounds, we tested the binding of six compounds (iKeap1, 2, 7, 8, 9 and 22) to KEAP1 by a suite of NMR-based ligand-detected experi-ments. Out of these six compounds, five are displacers and one of them (iKeap9) is a binder. These six compounds were selected on the basis of the solubility constraints of the NMR experiments, the SPR Kd value, and/or their ability to displace the peptide. We used differential line broadening (DLB), saturation transfer difference (STD), Car-Purcell-Meiboom-Gill (CPMG)-based transverse relaxation time experiments, and protein-observed 1H-13C heteronuclear multiple-quantum cor-relation (HMQC) experiments to confirm binding of the compounds to KEAP1. The ligand-detected NMR experiments confirmed that all six of the tested SPR Level-3 active compounds bind to KEAP1 (Fig.3, Extended Data Figs.7, 8). Protein-detected 1H-13C HMQC experiments show that the compounds engage KEAP1 in a specific manner, at the targeted NRF2 binding site. In the absence of resonance assignments, we use the fact that the compounds perturb a subset of KEAP1 reso-nances affected by the addition of the NRF2 peptide as evidence for competitive binding. These compounds are shown in Supplementary Figs.1, 2, and 3. Details about the other active compounds is provided in Supplementary Information SectionB.
Two of our top hits, iKeap1 and iKeap2 are able to displace the NFR2 peptide from KEAP1. Both of the compounds are predicted to engage the NRF2 binding pocket on KEAP1, located at the entrance to the tun-nel formed by the β-barrel. (Fig.3a, b). In comparison to iKeap2, iKeap1 descends deeper into this central tunnel of KEAP1. SPR results showed that iKeap1 and iKeap2 bind to KEAP1 with a binding affinity of 114 nM and 158 nM, respectively (Fig.3c, d). NMR-based ligand-detected experi-ments confirmed that both iKeap1 and iKeap2 directly bind to KEAP1 (Fig.3e, f). FP assays showed that iKeap1 is able to displace NRF2 peptide with an IC50 of 258 nM and iKeap2 displaces the NRF2 peptide with an IC50 of 2.7 µM (Fig.3g, h). BLI measurements additionally confirmed that both iKeap1 and iKeap2 are able to displace the NRF2 peptide from KEAP1. iKeap1 exhibits similarity to a previously reported naphthalene-based compound with a lower IC50 (IC50 = 2.7 µM; compound C17 in Sup-plementary Table1 and Extended Data Fig.5d)25. C17 was identified as the best hit in a high-throughput screen (HTS) of 270,000 compounds25.
.../...
T O A C C E S S T H E R E S T O F T H E S T U D Y, P L E A S E V I S I T T H E S O U R C E .
.











