
































.
Highlights
• Cardiovascular cell senescence is distinct from quiescence.
• Senescent cardiovascular cells contribute to various disease development and cardiovascular ageing.
• Senolytics-induced apoptosis of senescent cardiovascular cells alleviates CVD onset and progression.
• Senescence immunotherapy is a promising strategy to target senescent cardiovascular cells.
Abstract
Cardiovascular disease (CVD) is the most common disease to increase as life expectancy increases. Most high-profile pharmacological treatments for age-related CVD have led to inefficacious results, implying that novel approaches to treating these pathologies are needed. Emerging data have demonstrated that senescent cardiovascular cells, which are characterized by irreversible cell cycle arrest and a distinct senescence-associated secretory phenotype, accumulate in aged or diseased cardiovascular systems, suggesting that they may impair cardiovascular function. This review discusses the evidence implicating senescent cells in cardiovascular ageing, the onset and progression of CVD, and the molecular mechanisms underlying cardiovascular cell senescence. We also review eradication of senescent cardiovascular cells by small-molecule-drug–mediated apoptosis and immune cell-mediated efferocytosis and toxicity as promising and precisely targeted therapeutics for CVD prevention and treatment.

1. Introduction
Human life expectancy is significantly increasing due to the better quality of water, food, hygiene, housing, and lifestyle, as well as vaccine usage and improved medical care (Foreman et al., 2018). As projected, the percentage of the global population of age ≥ 65 years will increase from 13% in 2010 to 19% in 2030, whereas those age ≥ 85 years will increase from approximately 0.03% in 2010 to approximately 1.4% in 2030 (Kontis et al., 2017). Advanced age has been well recognized as the leading unmodifiable risk factor for chronic fatal diseases (Niccoli and Partridge, 2012), including cardiovascular disease (CVD) (Shakeri et al., 2018), cancer, and neurodegenerative diseases (Baker and Petersen, 2018). Among these, CVD is the most common disease to increase globally as populations continue to age (Partridge et al., 2018). CVD is the leading cause of death in the elderly (Roth et al., 2017). However, the mechanisms underlying development of age-related CVD are largely unknown. Cellular senescence, a state of permanent cell-cycle arrest despite continued viability and metabolic activity, presents in diseased cardiovascular tissues and is strongly associated with cardiovascular ageing (Shakeri et al., 2018). Senescence is different from ageing, which is characterized by progressive functional decline. Senescence generally happens at the cellular level, whereas ageing occurs on the tissue or organ level. Cell senescence drives tissue ageing (McHugh and Gil, 2018) and is also different from cell quiescence characterized by reversible cell cycle arrest. Cell senescence and quiescence have distinct features and roles in the pathophysiology of CVD. Growing evidence indicates that senescent cardiovascular cells tightly trigger or exacerbate the onset and progression of numerous CVDs, including atherosclerosis (Childs et al., 2016), arterial stiffening (Schellinger et al., 2019), aortic aneurysms (Chen et al., 2016), (re)stenosis, myocardial fibrosis (Sawaki et al., 2018), and heart failure. Here, we discuss the unique features of senescent cardiovascular cells, molecular mechanisms underlying cardiovascular cell senescence, and emerging roles of senescent vascular cells in CVD initiation and progression. We also summarize whether and how senotherapy targeting elimination of senescent cardiovascular cells by senolytics or the immune system could be used to improve cardiovascular function with normal ageing-, disease-, or cancer therapy-induced damage, ideally resulting in healthy longevity (Campisi et al., 2019; Ovadya and Krizhanovsky, 2018; van Deursen, 2019).
2. Cellular senescence or quiescence and development of CVD
Senescent cardiovascular cells are especially abundant at sites of diseased or impaired cardiovascular systems, and accumulating evidence from human samples and mouse models demonstrates a causal role for senescent cells in the pathogenesis of age-related CVD, including atherosclerosis (Matthews et al., 2006), abdominal aortic aneurysm (AAA) (Chen et al., 2016), arterial stiffness (Roos et al., 2016), hypertension (Boe et al., 2013), and heart failure (Gude et al., 2018). We will review a body of work that, taken together, strongly suggests that cardiovascular cell senescence may have a significant role in the pathogenesis of CVD.
2.1. Cardiovascular cell senescence and quiescence
Cardiovascular cell senescence is defined as irreversible and permanent cell cycle arrest while cells remain metabolically active. Vascular cell senescence can be triggered by various detrimental stimuli, including but not limited to, radiation, oxidative stress, shortened telomeres (Matthews et al., 2006; Minamino et al., 2002), DNA damage, mitochondrial dysfunction, abnormal metabolism, and gene mutation. There are two kinds of vascular cell senescence (Bennett et al., 2016; Chi et al., 2019). The first is replicative senescence, irrevocable cell proliferation arrest after multiple cell divisions, which is generally mediated by telomere shortening (Kuilman et al., 2010). The second is stress-induced premature senescence (SIPS), a stable cell cycle arrest in the absence of any detectable telomere loss or dysfunction, which is usually induced by distinct endogenous or exogenous stresses (Kuilman et al., 2010). Cell senescence is a strategy used generally by mitotic cells to prevent dysregulated cell division. Emerging evidence demonstrates that cell senescence also occurs in post-mitotic cells, including cardiomyocytes and mature adipocytes (Sapieha and Mallette, 2018). In general, DNA damage in telomere regions drives post-mitotic cardiomyocyte senescence (Anderson et al., 2019). p53 induction mediates the senescence of post-mitotic adipocytes (Minamino et al., 2009). Upregulation of pro-senescence factor p21 triggers cell senescence in post-mitotic dopaminergic neurons (Riessland et al., 2019). Cardiovascular cell senescence is vital for the maintenance of cardiovascular tissue homeostasis during embryonic development, tissue regeneration, and wound healing (Demaria et al., 2014). However, persistent accumulation of senescent cells in cardiovascular tissues will impair cardiovascular function and has been implicated in the pathogenesis of age-related CVD. In contrast, cardiovascular cell quiescence with reversible cell cycle arrest usually occurs due to a lack of nutrition or growth factors (Blagosklonny, 2011).
2.1.1. Hallmarks of cardiovascular cell senescence
Senescent cardiovascular cells usually differ greatly from non-senescent cardiovascular cells, including proliferating cells and quiescent cells (Table 1). Senescent cardiovascular cells present several morphological and molecular features (Table 2) that may serve as suitable markers and therapeutic targets for these cells. Senescent cardiovascular cells generally display a characteristic flattened and enlarged morphology (Coleman et al., 2010; Meijles et al., 2017), increased senescence-associated beta-galactosidase (SA β-gal) activity (Matthews et al., 2006), telomere attrition, and accumulation of cyclin-dependent kinase inhibitor p16ink4a or p21 (Morgan et al., 2013). The prominent feature of senescent cardiovascular cells is the senescence-associated secretory phenotype (SASP). Senescent vascular cells secrete a variety of pro-inflammatory cytokines (e.g. IL-6, IL-8), growth factors (e.g. vascular endothelial growth factor [VEGF], platelet-derived growth factor AA [PDGF-AA]) (Demaria et al., 2014), chemokines, and matrix metalloproteinases (MMPs). Senescent vascular cells exhibit a SASP that enables them to communicate with other cells, as well as the microenvironment, and to promote the senescence of neighboring cells, tissue regeneration, and embryonic development (Munoz-Espin et al., 2013). A critical feature of senescent cells is that they are more resistant than non-senescent cells to both extrinsic and intrinsic pro-apoptotic stimuli, which may be due to the transcriptional and cap-independent translational upregulation of pro-survival BH2 family proteins (BCL-W, BCL-XL, and BCL-2) (Yosef et al., 2016). Another surrogate marker of vascular cell senescence is the induction of telomere-associated foci (TAF) of DNA damage (Roos et al., 2016). DNA methylation may function as a biomarker for vascular cell senescence and biological ageing (Field et al., 2018).

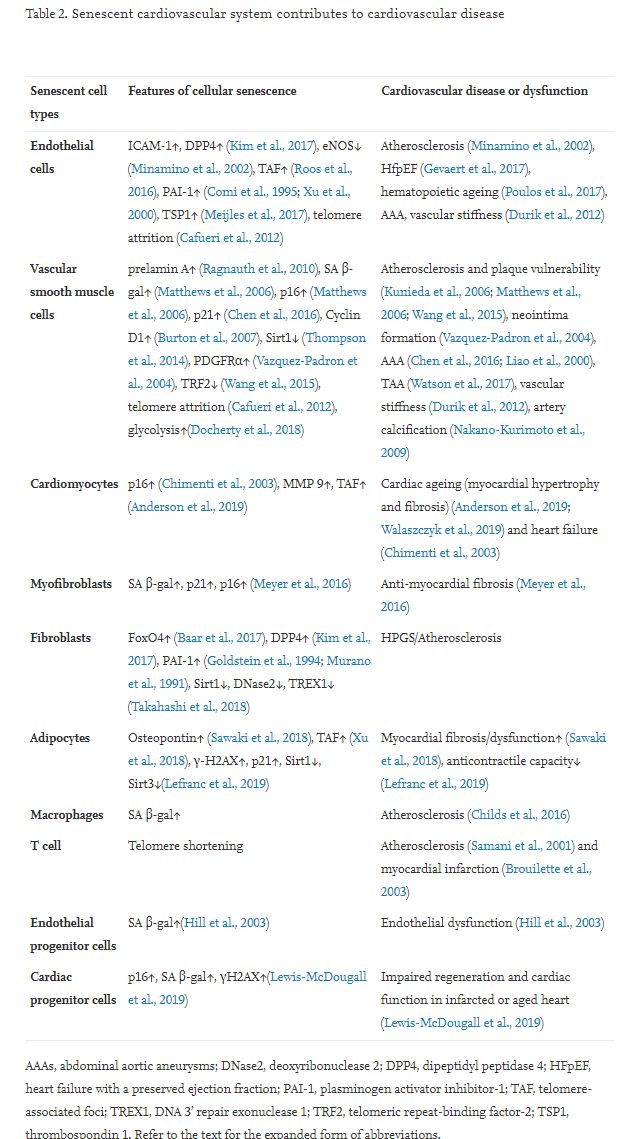
Notably, one type of cardiovascular cell may have its unique senescent hallmarks with different kinds of senescence. For example, passaged vascular smooth muscle cells (VSMCs) exhibit p16, but not p21, elevation in replicative senescence, whereas p21, but not p16, is expressed in oxidative SIPS (Matthews et al., 2006). Endothelial cell (EC) SENEX is upregulated in SIPS, but not in replicative senescence (Coleman et al., 2010). Upregulation of fibroblast senescence marker dipeptidyl peptidase 4 (DPP4, also known as CD26) is much stronger in replicative senescence than in ionizing radiation (IR)-induced premature senescence (Kim et al., 2017). Middle-aged wild-type lung ECs show elevation of p53 and p21, but not p16, compared with younger counterparts (Meijles et al., 2017). Cyclin D1 reactivity (upregulation) is a more accurate marker than SA β-gal activity for replicative senescence in human VSMCs (Burton et al., 2007). Thrombospondin 1 (TSP1) protein levels are increased in senescent ECs, but not in VSMCs (Meijles et al., 2017). Thus, different cardiovascular cells have distinct molecular signatures of senescence, which may serve as potential therapeutic targets for selective elimination of different senescent cells.
2.1.2. Features of cardiovascular cell quiescence
Most cardiovascular cells in a healthy adult are quiescent (Eelen et al., 2018). Quiescent cardiovascular cells are characterized by reversible cell cycle arrest at G0 (Kalucka et al., 2018) and responsiveness to external stimuli, including both growth factors and apoptotic agents, which is distinct from senescent cells (Table 1). Different cardiovascular cells may have unique features of quiescence. EC quiescence has been well studied. Generally, Notch signaling induces endothelium quiescence (Harrington et al., 2008), which increases fatty acid β-oxidation (FAO) via elevation of Notch1-mediated carnitine palmitoyltransferase 1A (CPT1A) up to levels 3- to 4-fold greater than in proliferating ECs to sustain the tricarboxylic acid cycle for redox homeostasis through regeneration of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH). Quiescent ECs also have upregulated endothelial nitric oxide synthase (eNOS) and prostaglandin G/H synthase 1 (PTGS1), as well as downregulated glycolysis (Kalucka et al., 2018). Also, forkhead box O1 (FoxO1) activation enhances EC quiescence by downregulating Myc protein levels and triggering consequent glycolysis inhibition, whereas FoxO1 activation does not induce EC senescence and apoptosis (Wilhelm et al., 2016). FoxO1 activation also mediates quiescence of pulmonary artery smooth muscle cells (Savai et al., 2014). Supplementation with acetate (metabolized to acetyl-coenzyme A) restores endothelial quiescence and counters oxidative stress-mediated EC dysfunction in EC-specific CPT1A-deleted mice (Kalucka et al., 2018), offering therapeutic opportunities. Quiescent ECs stimulated by β-hydroxybutyrate (β-HB) present upregulated Oct4 and Lamin B1 (Han et al., 2018). Bone morphogenetic protein-9 (BMP9) can function as a vascular (endothelial) quiescence factor (David et al., 2008).
2.2. Cellular senescence contributes to CVD
Dysregulation of cardiovascular cell senescence is tightly linked to many human CVDs, such as heart failure, coronary artery disease, atherosclerosis, aortic aneurysm, and vessel (re)stenosis. The role of senescent cardiovascular cells in the etiology of these pathologies was recently established. It was reported that p16-positive cells are major drivers of the age-related cardiac phenotype that results in decreased lifespan in mice (Baker et al., 2016). Removal of senescent cells with p16 promoter activity inhibits both atherosclerotic plaque onset and progression and enhances plaque stability (Childs et al., 2016).
2.2.1. Endothelium senescence and CVD
ECs line the inner vascular wall, and their phenotype, fate, and function alternately depend on the organs and tissues in which they reside and the niches. Not only do ECs form the barrier of vessel walls, they also communicate via signals with neighboring cells to promote tissue regeneration and growth, as well as to control low-density lipoprotein (LDL) transcytosis and consequent atherogenesis (Huang et al., 2019). EC senescence is tightly linked to EC dysfunction (Kim et al., 2018b) and subsequent CVD development and progression (Table 2) (Bochenek et al., 2016; Pantsulaia et al., 2016; Regina et al., 2016). Minamino et al. first demonstrated that senescent ECs with strong SA β-gal activity are present in atherosclerotic lesions of human coronary arteries (Minamino et al., 2002). Atherosclerotic ECs have shortened telomeres compared with the ECs in the normal vessel wall (Ogami et al., 2004). ECs from the aneurysmal region also present a senescent phenotype with shorter telomeres and more severe oxidative DNA damage (Cafueri et al., 2012). Importantly, in a mouse ageing model, EC senescence contributes to heart failure without systolic dysfunction, specifically heart failure with preserved ejection fraction (HFpEF), that occurs in approximately 50% of all patients with heart failure (Gevaert et al., 2017). Also, EC senescence mediates thrombosis (complete vena cava occlusion) via elevation of plasminogen activator inhibitor-1 (PAI-1), an established marker and key mediator of cellular senescence (McDonald et al., 2010). EC premature senescence due to sirtuin deacetylase 1 (Sirt1) inhibition (Ota et al., 2007; Zu et al., 2010) may reversibly lead to vascular ageing and age-related decrease in exercise endurance (Das et al., 2018). Senescence of bone ECs (type H ECs with high expression of CD31 and endomucin) may trigger dysfunctional vascular niches for hematopoietic stem cells (Kusumbe et al., 2016), which may accelerate atherosclerosis development in mice (Fuster et al., 2017).
2.2.2. Senescence of vascular smooth muscle cells and CVD
VSMC senescence is profoundly associated with and contributes to numerous CVDs, including atherosclerosis (Bennett et al., 2016; Gardner et al., 2015; Grootaert et al., 2018), aortic aneurysm (Cafueri et al., 2012), and fibrotic neointima formation (Komaravolu et al., 2019). VSMCs from aged thoracic aortas express higher levels of platelet-derived growth factor receptor-alpha (PDGFR-α) and are resistant to apoptosis induced by serum starvation or nitric oxide (Vazquez-Padron et al., 2004). VSMCs derived from human atherosclerotic plaques have a lower level of proliferation compared with cells from the regular arterial media, suggesting that plaque VSMCs are prematurely senescent (Bennett et al., 1998). Human plaque VSMCs are characterized by higher p16 and p21 expression, hypophosphorylation of retinoblastoma (RB), stronger SA β–gal activity, and sizeable flattened cell morphology, when compared with normal VSMCs (Gorenne et al., 2006). Matthews et al. reported that senescent VSMCs are present in the fibrous cap of human advanced carotid atherectomies (Matthews et al., 2006), and VSMCs within the fibrous cap demonstrate remarkable telomere loss compared with medial VSMCs of the same lesion. Furthermore, telomere shortening of intimal VSMCs is tightly linked to increasing severity of atherosclerosis (Matthews et al., 2006). Angiotensin II (Ang II) has been reported to accelerate the development of atherosclerosis via induction of premature senescence by the p53/p21-dependent pathway in VSMCs, but not bone marrow cells (Kunieda et al., 2006). VSMC senescence due to Sirt1 inactivation increases atherosclerosis (Gorenne et al., 2013). Also, VSMC senescence contributes to plaque vulnerability, leading to myocardial infarction and stroke (Wang et al., 2015). VSMC-specific TRF2 overexpression in apolipoprotein E knockout (ApoE-/-) mice prevents senescence and consequently improves several features of plaque vulnerability (Wang et al., 2015).
Medial VSMCs derived from patient AAAs demonstrate accelerated replicative senescence compared to VSMCs from the corresponding adjacent (non-aneurysmal) inferior mesenteric artery of the same patient (Liao et al., 2000). Ang II induces VSMC senescence and resultant AAA formation via Sirt1 reduction (Chen et al., 2016). Medial VSMC senescence due to NAD+ reduction by inhibition of the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT) leads to human thoracic aorta (ascending aorta) aneurysm (Watson et al., 2017). VSMC senescence in the aorta also increases vascular stiffness (Durik et al., 2012). VSMC senescence induced by nicotine (Suner et al., 2004) may drive nicotine-mediated aortic and arterial stiffness (Ding et al., 2019). Replicative senescence of VSMCs instigates age-related medial artery calcification that is not concomitant with lipid or cholesterol deposit via runt-related transcription factor-2 (RUNX-2)-mediated osteoblastic transdifferentiation (Nakano-Kurimoto et al., 2009). Ageing exacerbates neointimal formation by wire injury in carotid arteries in mice (Vazquez-Padron et al., 2004). However, it is unknown if age-enhanced neointimal formation is due to VSMC senescence.
2.2.3. Immune cell senescence in CVD
Immune cell senescence (immunosenescence) plays a pivotal role in CVD initiation and progression (Alpert et al., 2019; Yu et al., 2016). Macrophages are the primary type of immune cell that plays critical roles in CVD development. Employing CD11b-driving diphtheria toxin (DT) receptor (DTR) transgenic mice, Stoneman et al. showed that monocyte/macrophage content positively contributes to atherosclerotic plaque development, collagen content, and necrotic core formation. However, monocyte reduction has minor effects on the established plaques (Stoneman et al., 2007). Mouse ageing is associated with accumulation of senescent macrophages that can be induced in young mice by senescent fibroblasts (Hall et al., 2016). Senescent macrophages accumulate in the sub-endothelial space during early atherogenesis (Childs et al., 2016). In advanced atherosclerotic plaques, senescent macrophages promote features of plaque instability, including diminished collagen content, elastic fiber fragmentation, and fibrous cap thinning, in descending aorta and brachiocephalic artery, by elevating MMP3 and MMP13 formation. Interestingly, selective removal of these p16-positive senescent cells without interfering with the senescence program by genetic or pharmacological strategies reverses atherosclerosis in mice (Childs et al., 2016). It was reported that older persons (over the age of 60 years) with the senescent marker of shorter telomeres in leukocyte DNA have a 3.18-fold higher mortality rate from heart disease (Cawthon et al., 2003), implying that senescent immune cells may lead to heart disease. Accelerated telomere shortening also presents in leukocytes of patients with severe coronary artery disease (Samani et al., 2001) and myocardial infarction (Brouilette et al., 2003). Plasmacytoid dendritic cells (pDCs, uniquely produce type I interferon) and regulatory T cells (Tregs) are concomitantly induced and co-localized in mouse atherosclerotic intima (Yun et al., 2016). Although the accumulation of intimal DCs increases in aged mice with accelerated atherogenesis (Liu et al., 2008), the causal function of senescent DCs and T cells in CVD development remains an unmet challenge. Recently, it was reported that human carotid artery plaques contain immune cells, including CD4+ or CD8+ T cells, natural killer (NK) cells, and macrophages (Fernandez et al., 2019). However, it is totally unknown whether the patient’s plaque immune components are senescent and the role of senescent immune components in human atherogenesis.
2.2.4. Senescent myofibroblasts and fibroblasts in CVD
Senescence of cardiac myofibroblasts is increased in perivascular fibrotic areas after transverse aortic constriction (TAC) compared with the sham-treated heart. Inhibition of premature senescence by genetic deletion of both p53 and p16 leads to enhanced fibrosis and cardiac dysfunction after TAC compared with the wild-type control heart. In contrast, induction of premature senescence by cardiac-specific adeno-associated virus serotype 9 (AAV9) (Suckau et al., 2009) gene transfer-mediated expression of cysteine-rich angiogenic inducer 61 (CYR61) (Jun and Lau, 2010) results in an approximately 50% reduction of perivascular fibrosis and improved cardiac function after TAC (Meyer et al., 2016). These data imply that premature senescence of myofibroblasts functions as an essential anti-fibrotic mechanism and is a promising therapeutic target for myocardial fibrosis (Condorelli et al., 2016). The roles and regulation of senescent fibroblasts and myofibroblasts in the development of CVD, including AAA, cardiac fibrosis, and arterial stiffness, warrant further investigation.
2.2.5. Senescence of vascular stem/progenitor cells and CVD
Ageing is frequently associated with dysfunction of stem or progenitor cells. Although cellular senescence of progenitor cells (PCs) contributes to multiple diseases (Nicaise et al., 2019), senescence of cardiovascular PCs in CVD progression has been less investigated. Circulating endothelial progenitor cells (EPCs) from human subjects at high risk for cardiovascular events or older subjects have higher percentages of in vitro senescence (Hill et al., 2003) or functional impairment (e.g. decreased migration and proliferation) (Heiss et al., 2005), which is correlated with vascular or EC dysfunction, a key trigger of atherogenesis. Depletion of growth differentiation factor 11 (GDF11) or telomerase reverse transcriptase (TERT) causes senescence of young VEGFR2+/CD133+ EPCs, leading to impaired vascular function and angiogenesis in vitro and in vivo (Zhao et al., 2019). However, it is unknown whether EPC senescence contributes to the onset and progression of CVD.
Although the endogenous cardiomyocyte renewal capacity of adult cardiac stem/progenitor cells (CSCs/CPCs) still matters of debate (van Berlo et al., 2014; Vicinanza et al., 2018), they exert a beneficial effect on cardiac function in animal models of cardiac ischemic injury (Vagnozzi et al., 2020). Age affects the senescence of human CSCs from older patients (Lewis-McDougall et al., 2019; Nakamura et al., 2016), and it also enhances mouse CSC senescence (Torella et al., 2004). Indeed, c-kit+ cardiac CPCs from aged (24 months) C57BL/6 mice have increased senescent phenotype, decreased stemness, and impaired ability to upregulate paracrine factors for angiogenesis (Castaldi et al., 2017). Overall, CSC senescence mediates cardiac ageing and heart failure (Cianflone et al., 2019; Torella et al., 2004). Interestingly, elimination of senescent CPCs using dasatinib + quercetin (D + Q) senolytics attenuates the SASP and its effect on promoting senescence of healthy non-senescent CPCs in vitro. Moreover, systemic ablation of senescent cells in aged mice in vivo using senolytics (D + Q) leads to resident CPC activation and enhanced heart regenerative capacity (Lewis-McDougall et al., 2019). Ageing induces senescence of cardiac mesenchymal stem cells (MSCs) associated with decreased CD90 expression, resulting in impaired EC differentiation potentials and enhanced SASP (Martini et al., 2019), which may contribute to cardiac disease. Additionally, CVD risk factors, such as type 2 diabetes, depletes circulating pro-vascular PCs characterized by high aldehyde dehydrogenase activity and CD34+ (Terenzi et al., 2019). Importantly, in patients with their first acute myocardial infarction, tight glycemic control reduces senescent myocyte precursor cells, thus increasing the regenerative potential of the ischemic myocardium (Marfella et al., 2012).
3. Molecular mechanisms of cardiovascular cell senescence
There are multiple mechanisms involved in cardiovascular cell senescence. Here, the review summarizes several key underlying molecular mechanisms.
3.1. Progeria and vascular cellular senescence in cardiovascular ageing and diseases
The homeostasis of the cell nucleus is profoundly modified during cellular senescence. Defects of the nuclear lamina have been associated with several different diseases of accelerated ageing, including Hutchinson-Gilford progeria syndrome (HGPS) (Gonzalo et al., 2017; Gordon et al., 2014), mandibuloacral dysplasia (Novelli et al., 2002), and atypical Werner syndrome (Bonne and Levy, 2003). HGPS is an ultra-rare, early-onset, and severe genetic disease of premature ageing caused by a point mutation (C1824 T) in Lmna (G608 G) or Zmpste24 that disrupts nuclear lamin A processing, leading to the formation of mutated (truncated and farnesylated) prelamin A, generally referred to as progerin (50 amino acids deleted from the tail of prelamin A) (Kim et al., 2018a; Lee et al., 2016). Prelamin A elevation is linked to oxidative stress-mediated reduction of the lamin A-processing enzyme Zmpste24/FACE1 (Fig. 1) (Ragnauth et al., 2010). HGPS patients exhibit severe premature arteriosclerosis characterized by VSMC calcification and attrition, as well as prominent adventitial fibrosis, and die in their early teens (younger than 15 years), mainly due to myocardial infarction or stroke (Olive et al., 2010).

Fig. 1. Prelamin A accumulation leads to vascular cell senescence and multiple cardiovascular diseases. ┴, inhibits. Refer to the text for the expanded form of abbreviations.
Prelamin A accumulation in multiple cardiovascular cells contributes to their senescence. For example, senescent VSMCs rapidly accumulate prelamin A and present defective nuclear morphology in vitro, both of which are reversible by treatment with farnesylation inhibitors and statins (Fig. 1) (Ragnauth et al., 2010). In human arteries, prelamin A does not accumulate in young and healthy vessels but is prevalent in medial VSMCs from aged individuals or in atherosclerotic lesions, where it often colocalizes with senescent and degenerative VSMCs. Knockdown of FACE1 recapitulates the prelamin A-induced defects in nuclear morphology in aged VSMCs, whereas prelamin A overexpression promotes VSMC senescence through disrupting mitosis and inducing DNA damage in VSMCs, leading to premature senescence (Ragnauth et al., 2010). Selective overexpression of progerin in VSMCs, but not macrophages, leads to VSMC loss and promotes LDL retention in the aorta and the resultant atherogenesis and death in a mouse model of HGPS (Hamczyk et al., 2018). Disruption of the linker of the nucleoskeleton and cytoskeleton (LINC) complex in VSMCs ameliorates progerin-induced VSMC apoptosis and limits the accompanying adventitial fibrosis (Kim et al., 2018a). Furthermore, VSMC-derived progerin accelerates atherogenesis via inducing endoplasmic reticulum (ER) stress in the aorta (Hamczyk et al., 2019). Mice with progerin overexpression in ECs (progerinecTg) develop perivascular and cardiac fibrosis, cardiac hypertrophy (Fig. 1), and premature death without VSMC depletion (Osmanagic-Myers et al., 2019). Also, progerin expression is increased in human hearts with dilated cardiomyopathy and is strongly associated with left ventricular remodeling and myocardial ageing (Messner et al., 2018). Left ventricular diastolic dysfunction is the most prevalent echocardiographic abnormality in HGPS patients, and its prevalence increases with age (Prakash et al., 2018). Recently, Beyret and colleagues employed a single-dose systemic administration of AAV9-delivered CRISPR-Cas9 components with lamin A/progerin reduction via facial vein injection to repress HGPS in a mouse model (Beyret et al., 2019). At the same time, another group using intraperitoneal injection of AAV9-mediated CRISPR-Cas9 to ameliorate HGPS in LmnaG609G/G609G mice (Santiago-Fernandez et al., 2019). All the results indicate that prelamin A accumulation in different cardiovascular cells due to impaired lamin A processing is a novel biomarker of cardiovascular ageing and contributes to CVD development (Fig. 1) and therefore represents a novel therapeutic target to ameliorate the effects of age-induced cardiovascular dysfunction.
3.2. Impaired autophagy leads to cardiovascular cell senescence
Autophagy is a “housekeeping” cellular process recognized as a mechanism for cell survival when cells encounter stress, including nutrient deprivation or hypoxia, in which cells degrade their dysfunctional proteins, macromolecules, or sub-organelles in lysosomes and recycle them to produce the required raw materials for biosynthesis or energy generation (Anding and Baehrecke, 2017; Grootaert et al., 2018). In general, autophagy appears to be constitutively active in the cardiovascular system, but activity decreases with age (Kroemer, 2015; Shirakabe et al., 2016). Importantly, inhibited general autophagy or special autophagy of mitochondria (mitophagy) leads to or accelerates cardiovascular ageing (Abdellatif et al., 2018). Dysfunctional autophagy in ECs, VSMCs, and macrophages plays a detrimental role in atherogenesis (Fig. 2). Growing evidence implies that decreased autophagy results in cardiovascular cell senescence (Sasaki et al., 2017). For instance, VSMC-specific deficiency of the essential autophagy factor autophagy-related 7 (ATG7) causes accumulation of SQSTM1/p62 and accelerates SIPS. ATG7 deletion in VSMCs of ApoE-/- mice promotes ligation-induced neointima formation and Western diet-induced atherogenesis in mice (Grootaert et al., 2015). Interestingly, moderate activation of autophagy by rapamycin has been shown to repress VSMC replicative senescence (Tan et al., 2016) and stabilize progressed atherosclerotic plaques (Luo et al., 2017). Inhibition of autophagic adaptor p62-mediated selective autophagy stabilizes and increases GATA4 protein, which initiates and maintains the SASP, thus triggering senescence in fibroblasts (Kang et al., 2015).

Fig. 2. Defective autophagy and cardiovascular cell senescence. ┴, inhibits. Refer to the text for the expanded form of abbreviations.
3.3. Mitochondrial dysfunction causes cardiovascular cell senescence
Mitochondrial dysfunction usually drives cellular senescence (Chapman et al., 2019; Wiley et al., 2016), which is characterized by lower NAD+/NADH ratios (Mouchiroud et al., 2013; Watson et al., 2017; Wiley et al., 2016), excluding RAS oncogene-induced fibroblast senescence (Nacarelli et al., 2019). In general, mitochondrial fission reduction-caused inhibition of mitophagy contributes to senescence in multiple cell types by mitochondrial dysfunction (Fig. 3). For example, mouse heart with mitochondrial imbalance between fission (fragmentation) and fusion develops mitochondrial senescence and heart failure due to impaired mitophagy (Song et al., 2017). Furthermore, increased mitochondrial fission associated with elevation of mitochondrial reactive oxygen species (ROS), but not ER stress, triggers EC senescence and dysfunction, including impaired EC-dependent vasorelaxation and angiogenesis (Kim et al., 2018b). Kim and colleagues recently identified protein disulfide isomerase A1 (PDIA1) as a thiol reductase for the mitochondrial fission protein dynamin-related GTPase1 (Drp1) at Cys644. Diabetic reduction of PDIA1 induces Drp1 sulfenylation (oxidation) at Cys644, promoting Drp1 GTPase activity, which leads to mitochondrial fission contributing to EC senescence (Kim et al., 2018b). On the other hand, ageing also leads to mitochondrial dysfunction. For example, ageing elevates RNA-binding protein Pumilio2 (PUM2) in mouse muscle, which translationally downregulates mitochondrial fission factor (MFF, an outer mitochondrial membrane protein) and thereby inhibits mitochondrial fission and mitophagy, resulting in mitochondrial dysfunction (D’Amico et al., 2019). Interestingly, NAD+ replenishment restores defective mitophagy and mitochondrial function in fibroblasts and consequently restrains the accelerated ageing in Caenorhabditis elegans and Drosophila melanogaster models of Werner syndrome (Fang et al., 2019), a human premature ageing disease. It is unknown whether clearance of dysfunctional fragmented mitochondria by guanine derivative-targeted cargo-mediated mitophagy (Takahashi et al., 2019) attenuates cardiovascular cell senescence.
.../...
.
Edited by Engadin, 15 April 2020 - 10:56 PM.














