































.
S O U R C E : International Journal of Molecular Sciences
Abstract
Throughout life, organisms are exposed to various exogenous and endogenous factors that cause DNA damages and somatic mutations provoking genomic instability. At a young age, compensatory mechanisms of genome protection are activated to prevent phenotypic and functional changes. However, the increasing stress and age-related deterioration in the functioning of these mechanisms result in damage accumulation, overcoming the functional threshold. This leads to aging and the development of age-related diseases. There are several ways to counteract these changes: (1) prevention of DNA damage through stimulation of antioxidant and detoxification systems, as well as transition metal chelation; (2) regulation of DNA methylation, chromatin structure, non-coding RNA activity and prevention of nuclear architecture alterations; (3) improving DNA damage response and repair; (4) selective removal of damaged non-functional and senescent cells. In the article, we have reviewed data about the effects of various trace elements, vitamins, polyphenols, terpenes, and other phytochemicals, as well as a number of synthetic pharmacological substances in these ways. Most of the compounds demonstrate the geroprotective potential and increase the lifespan in model organisms. However, their genome-protecting effects are non-selective and often are conditioned by hormesis. Consequently, the development of selective drugs targeting genome protection is an advanced direction.
1. Introduction
The accumulation of genome damage and somatic mutations leading to genome instability are important determinants and hallmarks of aging [1,2,3]. Somatic mutagenesis as a key mechanism of aging was proposed by Leo Szilard in 1959 [4]. At the same time, recent theories also explain the nature of aging by impairments in maintaining the genome functioning stability (particularly, somatic mutation catastrophe theory) [5].
The consequences of the failure of mechanisms to maintain genome stability are vividly illustrated by the pathological patterns of numerous accelerated asging syndromes that are caused by mutations in DNA repair genes (for example, Werner, Cocaine, Bloom syndromes, xeroderma pigmentosum, ataxia-telangiectasia, and others) and nuclear architecture maintenance genes (laminopathy, in particular, Hutchinson–Gilford syndrome) [6,7,8,9,10]. On the other hand, an increased expression of a number of genes, providing a response to DNA damage and repair, causes an increase in the lifespan of model animals [2,11]. Species with extreme longevity, such as naked mole rats, Brandt bats, whales, mole rat Spalax, and parrots have adaptive features of repair mechanisms that increase the stability of their DNA [12,13,14,15,16]. In addition, reliable DNA protection is one of the reasons for the immortality of germline cells [17]. Genome instability accompanies age-related diseases such as cancer, heart failure, type 2 diabetes, chronic obstructive pulmonary disease, stroke, Alzheimer’s disease and Parkinson’s disease, chronic kidney disease, atherosclerosis, osteoporosis, sarcopenia [7,18].
Based on the foregoing, we suggest that stimulation of genome defense mechanisms may be a promising strategy to increase the lifespan and prevent the development of age-related diseases. There are several ways to achieve this goal: (1) prevention of DNA damage through stimulation of antioxidant and detoxification systems, as well as transition metal chelation; (2) regulation of DNA methylation, chromatin structure, non-coding RNA activity and prevention of nuclear architecture alterations; (3) improving DNA damage response and repair; (4) selective removal of non-functional and senescent cells (Figure 1). In the article, we have reviewed data about the genome-protecting effects of various trace elements, vitamins, polyphenols, terpenes, and other phytochemicals, as well as a number of synthetic pharmacological substances.
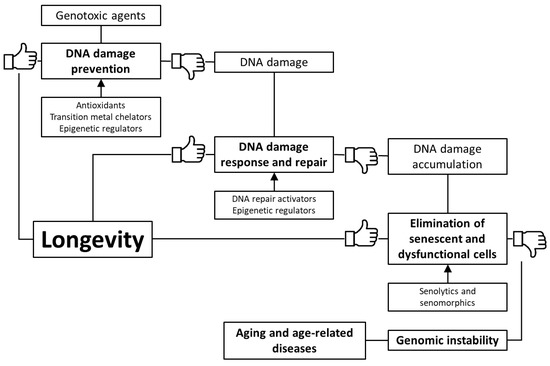
Figure 1. Key mechanisms of genome protection by pharmacological interventions.
2. Impairment of the Mechanisms for Maintaining Genome Stability during Aging
Throughout life, organisms are exposed to genotoxic dangers. Sources of DNA damage and mutagenesis are a variety of external factors (including physical and chemical agents, viral infections) and intracellular causes (spontaneous hydrolytic reactions, conversion of methylated cytosine to thymine, transposition of mobile genetic elements (MGEs), reactive oxygen species (ROS), DNA replication and DNA repair errors) [2]. Switching cells from glucose metabolism to β-oxidation also increases the level of DNA damage due to lipid peroxidation [19]. In addition, the depletion of the NAD+ pool [20] and insufficient synthesis of nucleotide DNA [21] cause aging. Lifestyle features, such as alcohol consumption [22], tobacco smoking [23], and a disturbance of circadian rhythms can also play a negative role [24].
During aging, the frequency of DNA damage and somatic mutations in tissues of animals and humans increases, genomic instability arises, which is expressed in a burst of point mutations, breaks, cross-linking of DNA strands, transpositions, translocations, aneuploidies [2]. Application of modern methods of analysis, in particular, single-cell genome sequencing [25] and transcript sequencing [26] allows seeing the somatic mutational landscape of the human body, including the age-dependent dynamics [27]. It is worth noting that different somatic cells accumulate mutations at different rates. As a result, clones of cells with a slightly different genotype are formed in an aging organism, forming somatic mosaicism [28,29,30]. This phenomenon is extremely widespread even among healthy people [31,32].
There are several levels of the cell protection against DNA damages and the accumulation of mutations, including scavenging of DNA-damaging molecules, repair of DNA damages, and elimination of dysfunctional cells from a dividing pool in response to permanent DNA damage through the initiation of cell senescence and apoptosis. In addition, maintaining the structure of chromatin, especially constitutive heterochromatin, plays an important role in ensuring the integrity and stability of genome functioning [18,33,34,35]. At a young age, compensatory mechanisms are activated to prevent phenotypic and functional changes. However, increasing stress and age-related impairment of the functioning of these mechanisms leads to the accumulation of damage, overcoming the functional threshold [36]. Dysregulation of these pathways can lead to accelerated or premature aging, age-related decline in the functional ability of vital organs, and the development of age-related diseases.
One of the basic mechanisms for preventing damage to cell macromolecules is the antioxidant defense system. Oxidative stress leads to an age-related increase in the cellular level of oxidatively modified macromolecules, including DNA, and this increase is associated with various pathological conditions, such as aging, carcinogenesis, neurodegenerative and cardiovascular diseases. This condition is counteracted by the antioxidant defense system, which includes enzymatic (superoxide dismutase, catalase, and glutathione peroxidase, and others) and non-enzymatic (vitamins A, C, E, thiols, flavonoids, and ubiquinones) [37]. The activity of antioxidant enzymes is significantly lower at an old age compared to young, while levels of free radicals and oxidative damage to DNA are increased [38,39]. In addition, a lack of antioxidant defense systems is observed in patients with ataxia-telangiectasia and Nijmegen breakage syndrome [40].
With age, there is a decrease in the catalytic activity of DNA repair proteins, including simple repair, base excision repair (BER) and nucleotide excision repair (NER), mismatch repair (MMR), repair of double-strand breaks (DSBR) by single-stranded annealing and the non-homologous end joining (NHEJ) (but not by homologous recombination (HR)). Such changes are combined not only with a reduced ability to quickly repair damaged regions but also with an increase in the frequency of repair errors because of impaired coordination of this process. For example, impaired BER coordination can cause the formation of inappropriate apurinic/apyrimidinic sites and single-stranded structures, especially under conditions of enhanced DNA damage [2]. In addition, somatic mutations in genes involved in DNA replication and repair can lead to a feedback loop of an exponentially increasing mutational load [5].
Genome stability is also determined by the state of constitutive heterochromatin. It covers a significant part of the genome and is represented by condensed, transcriptionally inactive DNA, consisting of a large number of nucleotide repeats. In particular, centromeric and telomeric regions belong to constitutional heterochromatin. It plays a critical role in providing mitosis, DNA replication, and repair, regulating gene expression and inhibiting the activity of MGEs [33,35]. The location of constitutive heterochromatin at the periphery of the nucleus has a protective function with respect to the coding DNA in euchromatin. In the nucleus, damaging agents are absorbed, blocked, and restored by constitutive heterochromatin, and its damaged DNA is removed and excluded from the nucleus into the cytoplasm through nuclear pore complexes [34]. In the case of viral infection, due to the mechanisms of maintaining heterochromatin, there is a long-term suppression of virus replication and gene silencing at the transcription level [35]. The accumulation of DNA damage during aging is probably associated with the age-related depletion and deregulation of heterochromatin. At the same time, an increase in the total amount of heterochromatin can contribute to improving the protection of genome and DNA coding proteins [35]. The loss of constitutive heterochromatin accompanies premature aging syndromes (in particular, Werner and Hutchinson–Gilford syndromes), mediates oncogenesis, and the development of cardiovascular diseases [33,35,41].
Recently, a number of studies have demonstrated links between genomic stability, metabolism, disease, and aging, which are mediated by the NAD+ levels and activity of NAD+-dependent enzymes, such as poly(ADP-ribose) polymerases (PARPs) [42,43] and sirtuins (class III histone deacetylases (HDAC)) [44,45]. NAD+ declining during aging contributes to the inactivation of sirtuins [46,47], which are involved in maintaining genomic stability due to coordination of DNA repair pathways [48,49], chromatin regulation [50], and telomere maintenance [51,52]. PARPs are considered as major NAD+-consuming enzymes during aging [46]. These proteins are recruited by DNA single-strand breaks and initiate repair processes by auto-ADP ribosylation, which utilizes NAD+ [53]. PARPs-mediated NAD+ consumption is enhanced during aging due to increased DNA damages [43], and inhibition of PARPs activity boosts NAD+ levels and SIRT1 activity [42,54,55,56]. Reduction, ablation or pharmacological inhibition of PARPs increase mitochondrial metabolism and boost mitochondrial respiratory capacity. At the organism level, these changes cause beneficial effects, in particular, protection from diet-induced obesity and enhance fitness [42,54,55].
In addition to SIRT1, other chromatin-modifying proteins such as SIRT6 and the heterochromatin protein HP1 undergo age-dependent changes. Their mutations in model animals lead to a shortened lifespan, while overactivation has a geroprotective effect [35,57]. SIRT6 is an important regulator of DNA repair enzymes and a chromatin modifier in response to DNA damage; its reduction plays a critical role in genomic instability [58]. Class I HDACs also decrease their activity during aging, which is especially pronounced in the brain [59,60,61]. These proteins are assembled into the nucleosome remodeling and deacetylation complex (NuRD), which is involved in the regulation of nucleosome position, and histone deacetylase activity and controls DNA damage response [60]. A member of this class, HDAC1, provides chromatin structure maintenance as well as is essential for DNA repair and replication processes [61,62]. At the same time, enhanced activation of classes I and II HDACs causes cancer and some other chronic diseases [62,63].
Various histone methyltransferases and demethylases can also coordinate the chromatin structure and the response to DNA damage. For example, these enzymes regulate the recruitment of DNA damage response proteins to DNA lesions and provide changes in gene transcription in response to genotoxic stress. Moreover, they can interact with non-histone proteins during the response to DNA damage [64].
The depletion of constitutive heterochromatin is closely associated with the telomere shortening. The role of telomere shortening in replicative senescence is well described. Replicative DNA polymerases are not able to fully replicate telomeres. In cells with constant renewal, including embryonic cells and stem cells, the telomerase enzyme is present. It consists of reverse transcriptase (TERT) and the RNA component of telomerase (TERC) and maintains telomere length by adding de novo telomeric repeats to the ends of newly synthesized chromosomes.
However, in somatic cells, telomerase in the nucleus is inactive, which leads to a cumulative loss of telomeric sequences during each division and leads to replicative senescence [7]. Telomeric dysfunction can be caused not only by the shortening of telomeres, but also by the disorder of their organization (imbalance in the formation of R-loops and guanine-quadruplexes) and by the formation of aberrant structures [65,66,67]. Abundant telomeric DNA damages contribute to genomic instability. In addition to the fact that telomeres are part of constitutive heterochromatin and are located on the periphery of the cell nucleus, their damage is not recognized by the corresponding sensors due to the presence of the shelterin complex [68,69]. In the cells of various mammalian organs, such damage accumulates, causing the formation of aging-related heterochromatic foci (SAHF) and activation of p16 [41,68,69]. In addition, TERT may be present in tissues with low replicative potential and perform non-canonical functions. It protects mitochondrial DNA from damage, maintains redox homeostasis, and protects cells from apoptosis [70,71,72].
Telomere length is not a key limiting factor in an organism lifespan [73]. This parameter varies in different tissues and cell types, and the telomere shortening rate changes over the course of an individual’s life [74,75]. At the same time, depleted telomeres are associated with an increased risk of all-cause mortality [76] and development of aging-dependent pathologies [74,77,78,79,80]. The loss of function of telomerase causes diseases characterized by premature aging, in particular, dyskeratosis congenita and its severe form, Hoyeraal–Hreidarsson syndrome [7,74,81].
As a result of the deficit of the repressive structure of constitutive heterochromatin, MGEs are activated [82,83]. They are widely represented in the eukaryotic genome (covering about 46% of the human genome; for example, Alu, LINE-1), but in the normal state, they are inactivated by transcriptional and post-transcriptional epigenetic mechanisms [84,85,86]. In the aging process, activation of MGEs occurs, which enhances genomic instability, provokes DNA damage, mutations, disruption, or change in the expression of normal genes [84,86,87].
In addition, the organization of the nuclear lamina affects the stability of the genome. A decrease in the amount of lamin B1, the accumulation of toxic levels of prelamin A and the expression of progerin (the pathogenic form of lamin A) lead to defects in the structure of the nucleus and are associated with cellular senescence and an organism aging [2,88,89]. Mutations in genes of a nuclear lamina cause premature aging syndromes called laminopathies (including Hutchinson–Gilford syndrome) [90,91]. It affects the speed of telomere shortening, the activity of genes and signaling pathways (including those associated with DNA damage response and aging), the organization of chromatin, and DNA methylation patterns [2,89]. In addition, the rigidity of the extracellular matrix through dysmorphia of the cell nucleus can provoke chromosome damages [92].
DNA damages induce a cell response that promotes the activation of signaling pathways that can drive various cell fates, including cellular senescence and apoptosis, mitochondrial dysfunction, hyperreactivity of innate immunity and inflammation [93,94,95,96].
Increasing genomic instability leads to a change in the transcription of vital genes, disruption of cellular metabolism, and causes cellular senescence. This leads to the accumulation of dysfunctional cells and genetic heterogeneity, a disruption of the regenerative potential, and physiological functions of tissues [3]. The consequences of the accumulation of DNA damages and somatic mutations are tissue-specific. In particular, the damage in macrophage DNA enhances inflammation [97], in neurons, it leads to cognitive impairment [98], in osteoprogenitor cells, it causes bone loss [99]. It is worth highlighting the accumulation of DNA damage and mutations in stem cells, as this influences their regenerative potential and creates a risk of tumor stem cells [100].
Tissue mechanisms also include a decrease in the ability of senescent cells to induce apoptosis [101] and a weakening of immunity that helps to eliminate them [102]. Cellular senescence is traditionally viewed as an irreversible cell cycle arrest that limits the proliferative potential of cells [103]. Senescent cells are involved in various physiological and pathological conditions, including tumor suppression, embryonic development, and tissue repair [104]. The senescent phenotype was described for postmitotic cells such as neurons [105], osteocytes [106], retinal cells [107], myofibrils [108] and cardiomyocytes [109].
The accumulation of senescent cells in various tissues is one of the hallmarks of aging [110] and the cause of age-dependent pathologies [111]. Cellular senescence contributes to the aging of the whole organism by reducing the regenerative potential of tissues (as a result of stem cell depletion) and through the induction of chronic inflammation (as a consequence of senescence-associated secretory phenotype (SASP) [112].
Resistance to apoptosis, in association with a decline in immune clearance, allows senescent cells to persist in the tissues for a long time, impairs tissue function, and underlies in age-related degenerative diseases, such as osteoarthritis, pulmonary fibrosis, atherosclerosis, diabetes, and Alzheimer’s disease [113]. Among the factors ensuring the resistance of senescent cells to apoptosis, ephrins (EFNB1 or 3), PI3Kδ, p21, BCL-xL, or plasminogen activator inhibitor-2 were identified [113,114].
Cellular senescence may be triggered by both external and internal stimuli [115]. External triggers arise from other senescent cells [116] and pro-inflammatory factors [117], inductors of cell proliferation (for example, growth hormone) [118], metabolic signals (for example, high glucose) [119], stress factors (for example, ionizing radiation) [120]. Internal triggers include replicative exhaustion [121] and telomere erosion [109], DNA damage [122], chromosomal instability [123], ROS [124], activation of oncogenes [125] and some other factors [93,115]. Persistent DNA damage response induces p21 and p16 cyclin-dependent kinase inhibitors and activation of the pRB retinoblastoma tumor suppressor pathway arresting the progress of the cell cycle [126,127].
.../...
.













