































.
O P E N A C C E S S S O U R C E : Cell Metabolism
SUMMARY
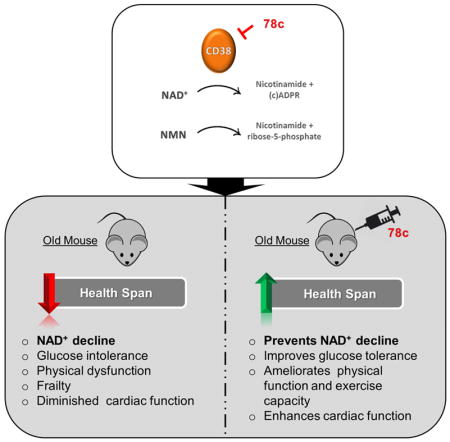
Aging is characterized by the development of metabolic dysfunction and frailty. Recent studies show that a reduction in nicotinamide adenine dinucleotide (NAD+) is a key factor for the development of age-associated metabolic decline. We recently demonstrated that the NADase CD38 has a central role in age-related NAD+ decline. Here we show that a highly potent and specific thiazoloquin(az)olin(on)e CD38 inhibitor, 78c, reverses age-related NAD+ decline and improves several physiological and metabolic parameters of aging, including glucose tolerance, muscle function, exercise capacity, and cardiac function in mouse models of natural and accelerated aging. The physiological effects of 78c depend on tissue NAD+ levels, and were reversed by inhibition of NAD+ synthesis. 78c increased NAD+ levels, resulting in activation of pro-longevity and health span-related factors including sirtuins, AMPK, and PARPs. Furthermore, in animals treated with 78c we observed inhibition of pathways that negatively affect health span, such as mTOR-S6K and ERK, and attenuation of telomere-associated DNA damage, a marker of cellular aging. Together, our results detail a novel pharmacological strategy for prevention and/or reversal of age-related NAD+ decline and subsequent metabolic dysfunction.
A reduction in nicotinamide adenine dinucleotide (NAD+) is associated with aging. XX et al show physiological and metabolic improvements of aging in old mice given with the small molecule 78c, which inhibits the NADase enzyme CD38. Mechanistically, mTORS6K/ERK and telomere-associated DNA damage pathways mitigate the NAD+ decline.
INTRODUCTION
NAD+ is a cofactor of key enzymes in glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation, participating in multiple redox reactions in cells. In addition, it serves as a substrate for several enzymes involved in cell signaling and DNA damage repair such as the sirtuins and poly (ADP-ribose) polymerases (PARPs) (Imai and Guarente, 2014; Verdin, 2015; Yoshino et al., 2017). The cellular NAD+ pool is controlled by a balance between the activity of NAD+-synthesizing and consuming enzymes (Aksoy et al., 2006; Bai et al., 2011; Bai et al., 2012; Barbosa et al., 2007; Grozio et al., 2013; Imai and Guarente, 2014; Mills et al., 2016; Nahimana et al.; 2009; Yang et al., 2007; Yoshino et al., 2011; Yoshino et al., 2017). Recent studies show that levels of NAD+ and its precursors decline during chronological aging and in progeroid syndromes, causing mitochondrial dysfunction and metabolic abnormalities (Braidy et al., 2011; Camacho-Pereira et al., 2016; Frederick et al., 2016; Gomes et al., 2013; Li et al., 2017; Massudi et al., 2012; Mills et al., 2016; Mouchiroud et al., 2013; Ramesey et al., 2008; Scheibye-Knudsen et al., 2014; Stein and Imai, 2014; Yoshino et al., 2011; Yoshino et al., 2017; Zhu et al., 2015). This decline in NAD+ levels is implicated in the development of age-related muscle dysfunction, glucose intolerance, and stem cell senescence (Cantó and Auwerx, 2012; Chini et al., 2016; Guarente, 2016; Imai and Guarente, 2014; Mills et al., 2016; Verdin, 2015; Schultz and Sinclair, 2016; Yoshino et al., 2017).
We recently reported that the NADase CD38 is one of the main enzymes responsible for age-related NAD+ decline in mammals, and that CD38 knockout (KO) mice are protected from this progressive deficit (Camacho-Pereira et al., 2016), indicating that CD38 may be an attractive target for the development of therapies to treat age-related metabolic dysfunction. Recently, thiazoloquin(az)olin(on)es, such as the small molecule 78c, have been identified as potent CD38 inhibitors (CD38i), (Becherer et al., 2015; Haffner et al., 2015). However, their mechanisms of action, specificity, and biological effects on age-related NAD+ decline and metabolic dysfunction have not been investigated. Here we demonstrate the potent anti-aging properties of the thiazoloquin(az)olin(on)e 78c. 78c reverses age-related NAD+ decline and ameliorates several metabolic, structural, and molecular features of aging in chronologically aged and progeroid mice. By kinetic analyses we show that 78c is highly potent and specific CD38i, with a reversible uncompetitive mechanism of action. We further demonstrate that 78c activates pro-longevity and health span-related factors, and inhibits those that negatively affect health span. In summary, these studies establish CD38 as a viable target for pharmacological NAD+-replacement therapy for age-related metabolic dysfunction.
RESULTS
78c is a potent, reversible, and uncompetitive inhibitor of CD38
We investigated the mechanism of inhibition of 78c and observed that 78c is a potent inhibitor of both human CD38 (recombinant, rhCD38) and murine CD38, with a Ki in the low nanomolar range (Fig. 1A–D). The inhibitory effect of 78c on CD38 NADase activity was reversible (Fig. S1A–B), and uncompetitive (Fig. 1B–C) decreasing both apparent Vmax and Km of CD38 for NAD+ (Fig. 1B). We confirmed this finding in assays using nicotinamide mononucleotide (NMN), a competitor of NAD+ and alternative substrate of CD38. Consistent with an uncompetitive inhibition model, NMN decreased the apparent Ki for 78c in a hyperbolic manner (Fig. S1C).
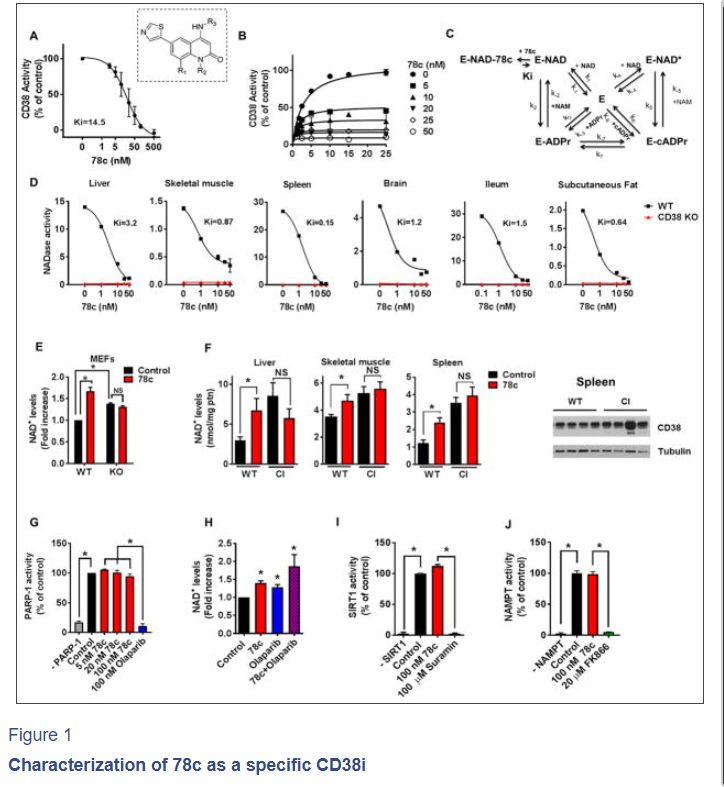
Figure 1. Characterization of 78c as a specific CD38i
(A) Hydrolase activity of recombinant human CD38 (rhCD38) in the presence of 78c, using 1,N6-ethenoadenine dinucleotide (ε-NAD+) as substrate (n=3 experiments, IC50 17.7 nM). Inset shows the structure of 78c. R1=H; R2=Me; R3=trans-4- OCH2CH2OMe-cyclohexyl.
(B) Effect of different substrate concentration on 78c inhibition of rhCD38 hydrolase activity. Experimental data were fitted by steady state equations derived from the kinetic model in panel
© Kinetic model of CD38 inhibition by 78c. Model depicts 2 interconnected cycles, one for the hydrolytic activity of rhCD38 (left side), and the other for the cyclase activity of the enzyme as well as the inhibition by its products nicotinamide and ADP- ribose (right side). In the reaction scheme, E represents the enzyme CD38; k(1-7), the substrate binding constants; Ki, the inhibitor binding constant.
(D) NADase activity in tissue homogenates from 1-year-old WT and CD38 KO mice, assayed in the presence of 78c (n=3 experiments). IC50: liver (3.8 NM), skeletal muscle (4.4 nM), spleen (0.7 nM), brain (14.8 nM), ileum (0.4 nM), subcutaneous fat (5.2 nM). AFU= arbitrary fluorescence units.
(E) NAD+ levels in WT and CD38 KO MEFs treated for 24 hours with 0.2 μM 78c (n=4–6 experiments). NAD+ levels were calculated relative to control WT MEF.
(F) NAD+ levels in tissues of 1-year-old WT and CD38 catalytically inactive (CI) mice treated with 78c or vehicle (Control) for 8 days (n=4 mice per group). Spleen protein lysates of WT and CI mice were immunoblotted for CD38 and Tubulin (right panel).
(G) Activity of human recombinant PARP1 in the presence of 78c or the PARP inhibitor olaparib (n=4 experiments).
(H) NAD+ levels in A549 cells treated for 24 hours with 0.5 μM 78c and/or 5 μM olaparib (n=3–6 experiments).
(I) Activity of human recombinant SIRT1 in the presence of 100 nM 78c or 100 μM SIRT1 inhibitor suramin (n=3 experiments).
(J) Activity of human recombinant NAMPT in the presence of 100 nM 78c or 20 μM of NAMPT inhibitor FK866 (n=3 experiments).
All values are mean ± SEM. *P < 0.05. NS=not significant. IC50=half maximal inhibitory concentration. See also Figure S1.
To refine our understanding of the mechanism of inhibition, we next tested the effect of the CD38 reaction products on the inhibition of CD38 by 78c. CD38 degrades NAD+ not only via its hydrolase activity, that results in the generation of the products nicotinamide (NAM), and ADP-ribose (ADPR), but also via its ADP-ribosyl cyclase activity, which generates the calcium signaling molecule cyclic-ADP-ribose (cADPR) (Malavasi et al., 2008). We observed that NAM decreased the apparent V0 and increased the Ki for 78c (Fig. S1D). In contrast, neither ADPR nor cADPR had significant effects on these kinetic parameters (Fig. S1E–F).
Next, we compared the effects of 78c on the hydrolase and cyclase activities of CD38. As shown in Fig. S1G, 78c was 10-fold less potent against the cyclase activity than the hydrolase activity of rhCD38 (Ki 100 ± 28 nM vs. 9.7 ± 1.5 nM, respectively). We further examined the effect of 78c on the activity of the ADP-ribosyl cyclase purified from the sea slug Aplysia californica (Malavasi et al., 2008). This enzyme is a pure cyclase that generates cADPR and NAM from the substrate NAD+ and has 25% homology to human CD38 (Malavasi et al., 2008). The Aplysia enzyme was not inhibited by 78c at concentrations up to 50 nM (Fig. S1H).
To further establish 78c as a specific inhibitor of CD38, we tested the effect of 78c on the CD38 homologous enzyme CD157 (Malavasi et al., 2008). The role of CD157 (also known as BST-1) in NAD+ metabolism in mammalians is not completely understood. In contrast to CD38, CD157 is a slow turnover enzyme that accepts nicotinamide riboside (NR) as its preferential substrate (Preugschat et al., 2014). Thus, we tested the effect of 78c on the catalytic activity of CD157 using both NAD+ and NR as substrates and observed no effect of 78c in the CD157 catalytic activities (Fig. S1I). Collectively, these studies establish that 78c is a specific, reversible, and uncompetitive CD38i that preferentially inhibits CD38 NADase activity.
78c increases NAD+ levels through inhibition of CD38 NADase activity
Previous studies have shown that a single oral dose of 78c increases tissue NAD+ levels in the liver of young mice fed a high fat diet (Becherer et al., 2015; Haffner et al., 2015). However, it is not known if this effect is dependent on the presence or enzymatic activity of CD38. We used mouse embryonic fibroblasts (MEFs) derived from wild type (WT) and CD38 KO mice to test whether CD38 expression is essential for the effect of 78c on NAD+ levels. We observed that WT MEFs have lower basal levels of NAD+ compared to CD38 KO MEFs (Fig. 1E and Fig. S1J). 78c nearly doubled the amount of NAD+ in WT MEFs, but had no effect on cellular NAD+ levels in CD38 KO MEFs (Fig. 1E).
Next, we tested if the NAD+ -modulating effect of 78c is dependent on CD38 catalytic activity in vivo. We generated a novel CD38 “knock-in” mouse model containing a mutation at E230 of the CD38 gene, which results in expression of a catalytically inactive (CI) CD38 (Fig. 1F and Fig. S1K–N). Although the CD38-CI animals expressed levels of CD38 similar to the WT mice (see Western blot in Fig. 1F and Fig. S1N), they had no detectable CD38 NADase activity and were protected against age-related NAD+ decline (Fig. S1L–M). We further observed that in contrast to CD38-CI mice, WT mice treated with 78c showed a significant increase in NAD+ levels in several tissues (Fig. 1F). Thus, we conclude that 78c-induced tissue NAD+ boosting is dependent on the inhibition of the catalytic activity of CD38.
78c is a specific inhibitor of CD38 and does not directly affect the activity or expression of other enzymes involved in NAD+ metabolism
It has been previously proposed that increases in cellular NAD+ levels can be pharmacologically induced by inhibition of NAD+ degradation catalyzed by enzymes such as Poly (ADP-ribose) polymerase (PARPs) (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014) or stimulation of enzymes involved in NAD+ synthesis such as NAMPT (Wang et al., 2014). Thus, to further characterize the mechanism by which 78c promotes increases in cellular NAD+ levels we investigated the effect of 78c in the activity and expression of key enzymes involved in NAD+ catabolism and anabolism.
PARP1 is the main PARP enzyme in mammalian tissues and utilize NAD+ during its catalysis (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014). Thus, it is very important to clearly demonstrate that 78c has no direct inhibitory or stimulatory effects on this enzyme. This is of particular importance since several studies have proposed that PARP1 inhibition can be used to promote increases in cellular levels of NAD+ by decreasing its degradation (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014). We first investigated if 78c had a direct effect on the enzymatic activity of PARP1 in vitro. While the PARP inhibitor olaparib completely inhibited the activity of this enzyme, 78c had no detectable effects on PARP1 activity (Fig. 1G). Next, we tested if inhibition of CD38 by 78c had an additive effect with PARP inhibition on cellular NAD+ levels. The PARP inhibitor olaparib increased cellular NAD+ levels by itself, and combination of olaparib with 78c produced an additive effect on the increase in cellular NAD+ levels we investigated the effect of 78c in the activity and expression of key enzymes involved in NAD+ catabolism and anabolism.
PARP1 is the main PARP enzyme in mammalian tissues and utilize NAD+ during its catalysis (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014). Thus, it is very important to clearly demonstrate that 78c has no direct inhibitory or stimulatory effects on this enzyme. This is of particular importance since several studies have proposed that PARP1 inhibition can be used to promote increases in cellular levels of NAD+ by decreasing its degradation (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014). We first investigated if 78c had a direct effect on the enzymatic activity of PARP1 in vitro. While the PARP inhibitor olaparib completely inhibited the activity of this enzyme, 78c had no detectable effects on PARP1 activity (Fig. 1G). Next, we tested if inhibition of CD38 by 78c had an additive effect with PARP inhibition on cellular NAD+ levels. The PARP inhibitor olaparib increased cellular NAD+ levels by itself, and combination of olaparib with 78c produced an additive effect on the increase in cellular NAD+ (Fig. 1H), demonstrating that the 78c-mediated increase in NAD+ occurs independent of PARP activity. Additionally, we tested if pharmacological inhibition of PARP with olaparib in vivo would increase NAD+ levels in WT and CD38 KO mice. There was an increase in NAD+ levels in tissues of WT animals treated with olaparib (Fig. S1O), and the NAD+-boosting effects of olaparib were not only preserved in tissues of CD38 KO mice such as liver and spleen, but appeared to be even potentiated in skeletal muscle (Fig. S1O). These data together demonstrate that 78c does not directly influence the activity of PARP, and that its NAD+-boosting effects are mediated by a mechanism that is independent of PARP activity.
Next we determined the effect of 78c on the activity and expression of other enzymes involved in NAD+ metabolism (Fig. 1I–J and Fig. S1P). We tested the effect of 78c on the NAD+-consuming enzyme SIRT1 (as a representative member of the sirtuin family), and on NAMPT (the rate-limiting enzyme in the salvage pathway of NAD+ synthesis) (Fig 1I–J). Using recombinant SIRT1 and NAMPT, we saw that 78c does not directly inhibit or stimulate these enzymes (Fig. 1I–J). In contrast, SIRT1 activity was decreased by the known SIRT1 inhibitor suramin (Fig. 1I), and NAMPT activity was completely abolished by its specific inhibitor FK866 (Fig. 1J). Furthermore, animals treated with 78c showed no significant changes in the expression of genes involved in NAD+ catabolism and anabolism (Fig. S1P). Altogether, these data clearly demonstrate that 78c increases tissue NAD+ levels by a mechanism dependent on the inhibition of the NADase catalytic activity of CD38, and not via its effect on other NAD+ metabolizing enzymes. Thus, our studies establish 78c as a potent and specific CD38i.
Metabolic function is improved in aged mice treated with 78c
Having established that 78c is a specific CD38 inhibitor that increases NAD+ levels, we next explored the potential role of 78c as a pharmacological therapy for aging-related metabolic dysfunction. Aging is characterized by the development of several metabolic changes that may limit health span (Huffman et al. 2016; Imai and Guarente, 2014; Justice et al., 2016; Verdin, 2016). For example, aging leads to an increased incidence of glucose intolerance, insulin resistance, and diabetes, as well as a decrease in physical activity and exercise capacity, and impairment in skeletal muscle architecture and cardiac function (Huffman et al., 2016; Justice et al., 2016). Many of these parameters are proposed as important endpoints for studies aiming to characterize potential health span-promoting interventions (Huffman et al., 2016; Justice et al., 2016). Of note, many of these phenotypes are regulated by NAD+ availability and the activity of NAD+-dependent enzymes (Camacho-Pereira et al., 2016; Gomes et al., 2013; López-Otín et al., 2016; Mills et al., 2016; Yoshino et al., 2011; Yoshino et al., 2017; Zhang et al., 2016).
Thus, we tested the effect of 78c on metabolic parameters in mice at different ages. As expected, aged mice (2-year-old) had lower tissue NAD+ levels, and worse glucose tolerance compared to younger mice (Fig. 2A–C and Fig. S2A–D). 78c promoted a significant increase in NAD+ levels in tissues of aged mice, but had a negligible effect on NAD+ levels of young mice (3-month-old) (Fig. 2A and Fig. S2C–D). Furthermore, after treatment with 78c, we noted an improvement in glucose homeostasis parameters in 2-year-old mice without changes in body weight, but no effects in glucose tolerance in either 3-month-old or 1-year-old mice (Fig. 2B–F and Fig. S2A–B). In support of these observations, we detected lower insulin levels in aged mice treated with 78c vs vehicle treated controls (Fig. 2D). The effect of 78c on glucose tolerance in these 2-year-old mice appears to be mediated, at least in part, by an increase in insulin sensitivity, as demonstrated by the HOMA-IR index and insulin sensitivity test (Fig. 2E–F). We further observed that 78c induced a decrease in expression of hepatic glucose-6-phosphatase, an enzyme involved in gluconeogenesis and endogenous glucose production in diabetes (Fig. 2G). Consistent with the decrease in glucose-6-phosphatase, we found that aged mice treated with 78c had lower blood glucose levels in response to administration of pyruvate, a precursor in the synthesis of glucose in the liver, suggesting suppression of the gluconeogenesis pathway by 78c (Fig. 2H). With the exception of increased expression of Pgc1α in muscle, no other significant changes in expression of genes related to mitochondrial biogenesis or glucose metabolism were observed in tissues of 78c treated mice (Figure 2G and Fig. S2E–G).
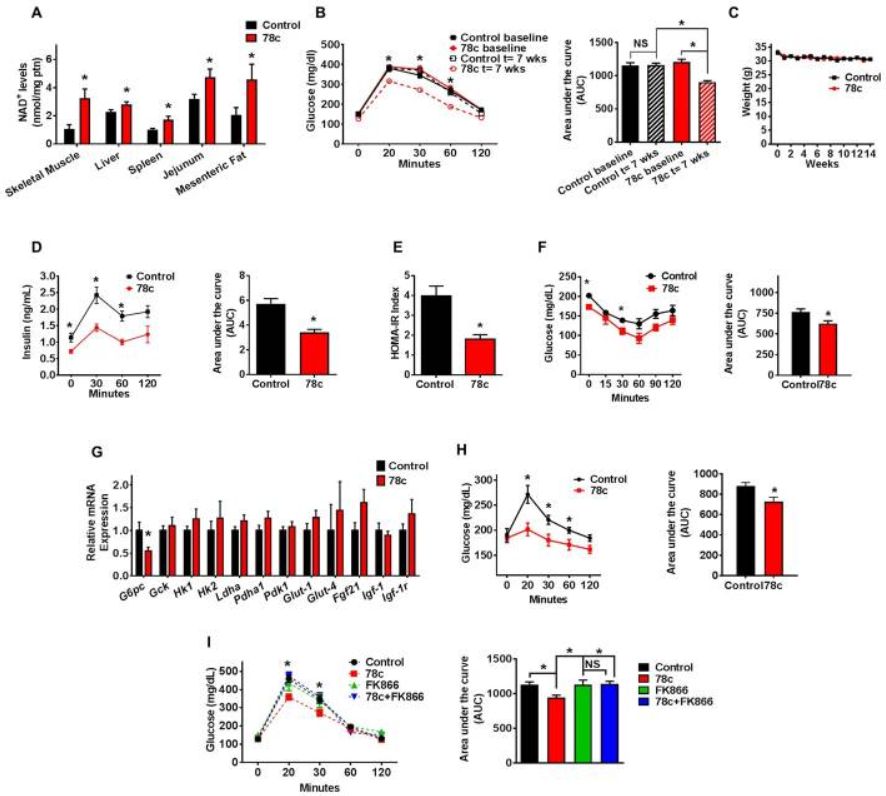
78c ameliorates metabolic dysfunction-associated features in chronologically aged mice
Aged (2-year-old) mice were treated with vehicle (Control) or 78c for up to 14 weeks.
(A) NAD+ levels in multiple tissues (n=4–13 mice per group) at the end of the treatment.
(B) Intraperitoneal glucose tolerance test (ipGTT) at baseline and after 7 weeks of treatment, and corresponding area under the curve (AUC) graph. Analysis by two-way repeated measures ANOVA with Bonferroni's post-tests shows significant interaction between the glucose curve for control and 78c-treated mice. Results in panels B and C show average of two independent experiments (n=18–24 mice per group).
© Weekly body weight measurements.
(D) Serum insulin measurement (ELISA) during ipGTT and corresponding AUC after 5 weeks of treatment (n=5 mice per group).
(E) Homeostatic model assessment index for insulin resistance (HOMA IR) (n=5 mice per group).
(F) Intraperitoneal insulin sensitivity test (ipIST) and corresponding AUC after 4 weeks of treatment (n=5 mice per group).
(G) Liver mRNA levels of glucose metabolism-related genes, determined by quantitative RT-PCR (n=6–19 mice per group) at the end of the treatment.
(H) Intraperitoneal pyruvate tolerance test (ipPTT) and corresponding AUC after 9 weeks of treatment (n=5 mice per group).
(I) ipGTT of 2-year-old mice after 4 weeks of treatment with vehicle (Control), 78c, FK866 (NAMPT inhibitor), or 78c+FK866 and corresponding AUC (n=5 mice per group). Statistical differences were determined using Two-way repeated measures ANOVA followed by multiple-comparison testing using Bonferroni’s post hoc analysis; *P < 0.05 Control compared with the 78c group. All values are mean ± SEM. *P < 0.05. NS=not significant. See also Figure S2.
.../...
.













