































.
O P E N A C C E S S S O U R C E : Drug Discovery Today
Highlights
• Multimorbidity is a common feature of old age and is accompanied by polypharmacy.
• Targeting the core processes of ageing offers a new drug discovery approach for multimorbidity.
• Inhibiting ageing processes reduces multiple diseases in mouse models.
• Current clinical trials with repurposed drugs have shown clinical benefit
• Targeting specific ageing processes with novel drugs may allow a more precise intervention.
Patients with multimorbidities have shorter life expectancy and their clinical management is more complex and expensive for healthcare systems currently focused on treating single diseases. Given that age is the major risk factor for multimorbidity, the challenge of treating these patients will only increase in coming years. Here, we review the case for targeting the core processes that drive the ageing phenotype as a novel pharmaceutical approach to multimorbidity. There is growing evidence that targeting ageing mechanisms can reduce or delay age-related diseases in animal models, and the first reports of clinical trials are now appearing. Although these trials currently focus on repurposed drugs, we propose several novel targets that would more specifically target ageing processes and thereby reduce multimorbidity and polypharmacy in future generations.
Introduction
Over the past 200 years, advances in public health and medical interventions have led to a dramatic increase in life expectancy, with current predictions suggesting that the total population of people aged over 60 years will double to approximately 2.1 billion by 2050 [1]. However, healthy life expectancy has not improved at the same pace, and so we are living longer but not healthier. For example, a global increase in life expectancy of 5 years between 2000 and 2015 was associated with only a 4.6-year improvement in healthspan [2]. In the UK, male life expectancy increased by 4.2 years between 1990 and 2010, but healthy life expectancy lagged behind, with a 2.7-year increase (for females, the equivalent data were 1.9 years and 1.1 years) [3]. As a result, an average of 16–20% of later life is spent in ill-health [4], with many individuals having age-related chronic diseases, such as arthritis, cancer, type 2 diabetes mellitus (T2DM), and cardiovascular and neurodegenerative disorders.
Crucially, older adults rarely experience just one of these conditions, and multimorbidity has become the norm in old age [5]. Multimorbidity, which is defined as the coexistence of two or more chronic conditions, is highly prevalent in high-income countries and is on the rise in low- and middle-income countries [5]. Multimorbidity affects at least 50 million people in the European Union [6], whereas 80% of Medicare users in the USA have at least two chronic conditions [7]. Although not limited to older adults, multimorbidity increases substantially with age, and is the main risk factor resulting in shorter life expectancy, worsening functional capacity, poorer quality of life, longer hospital stays, and increased healthcare costs for ageing populations around the world 8, 9. A cross-sectional study of 1.7 million people in Scotland found that 30.4% of people aged between 45 and 84 years, 64.9% of people aged between 65 and 84 years, and 81.5% of people aged 85 years or above had at least two chronic diseases [10]. Another notable statistic from this study is that multimorbidity onset can occur 10–15 years earlier in individuals living in more socially deprived areas [10]. Furthermore, every additional chronic disease in over 65 year olds shortens life expectancy by on average 1.8 years [7]. There is now mounting evidence that treatments effective in the single-disease context are far less so in the context of multimorbidity, and that polypharmacy leaves patients at serious risk of adverse effects 8, 11. The current challenge to overcoming the issue of multimorbidity is that medical practice, public health strategies, and drug discovery are still focused on treating these diseases individually, even though this no longer reflects the clinical reality for older patients and might even be harmful because of polypharmacy [11]. Thus, there is an urgent need for a novel approach to age-related multimorbidity, and we suggest that this requires a drug development and testing paradigm that focuses on the core processes driving multimorbidity rather than on its individual disease components. We suggest that it is vital that we move away from the one drug/one disease model for therapeutic interventions and drug discovery in multimorbidity towards a more holistic approach that brings its underlying causes into greater consideration. To achieve this, we need to better understand how multimorbidity develops, the specifics of different disease clusters in multimorbid patients, and the generic mechanisms driving each cluster.
Disease clustering in multimorbidity
Cross-sectional studies have begun to characterise patterns of associative multimorbidity, whereby specific pairings or clusters of diseases develop in individuals at a higher rate than expected from pure chance alone, in an attempt to provide useful evidence for better clinical management of patients with multimorbidities and to identify targetable pathways driving the different clusters [12]. Such studies have been varied in their choice of sample size, age setting, indices of multimorbidity, and statistical methodology, leading to inconsistent results that are difficult to compare between studies. Despite this variability, there does appear to be a growing consensus from multiple studies across different populations, as reviewed by Prados-Torres et al. [12], that multimorbidity patterns generally fall into three main clusters: cardiometabolic, neuropsychiatric, and musculoskeletal (Fig. 1). A cardiometabolic cluster, which was identified in ten of the 14 studies reviewed, most commonly involved at least two diseases, such as diabetes, hypertension, various types of heart disease (e.g., coronary heart disease or congestive heart failure), hyperlipidaemia, and obesity. A neuropsychiatric cluster was again identified in ten of the 14 cross-sectional studies reviewed and, in this instance, included at least one mental health condition. Of these, anxiety, depression, and chronic pain were the most frequent and were associated with a wide range of comorbidities, including, but not limited to, other neurological diseases (e.g., dementia, delirium, and Parkinson’s disease), respiratory problems [e.g., asthma or chronic obstructive pulmonary disease (COPD)], gastrointestinal conditions [e.g., gastro-oesophageal reflux disease (GORD) or liver disease], and musculoskeletal disorders (e.g., osteoporosis or arthritis). The musculoskeletal cluster included at least one musculoskeletal disorder, with the most common being arthropathy, back/neck pain, and osteoporosis. Similar to the neuropsychiatric cluster, a variety of comorbidities, such as obesity, and cardiovascular and neurological diseases, were commonly found within this cluster. These insights into the complex interactions of multiple diseases in multimorbidity are yet to be translated into clinical use for patient benefit. However, alongside more longitudinal explorations of multimorbidity trajectories to decipher when individuals are at risk of developing their first and/or next disease [13], clustering might hold the key to developing timely and targeted drug therapies for patients displaying the early stages of a specific multimorbidity cluster.

Figure 1. Disease clustering in multimorbidity. Cross-sectional studies recently identified patterns of associative multimorbidity, whereby specific pairings or clusters of disease develop in individuals at a higher rate than expected from pure chance alone. CAPITALS, major component; lower case, minor component. Abbreviations: CHD, chronic heart disease; CHR, chronic heart failure; COPD, chronic obstructive pulmonary disease; GORD, gastro-oesophageal reflux disease.
Targeting the potential generic drivers of multimorbidity: core ageing processes
Given that advancing age is the primary non-modifiable risk factor for developing many chronic diseases alone or concomitantly, a new paradigm, the ‘geroscience hypothesis’, recently emerged for preventing, delaying, or even reversing age-related diseases and loss of function 14, 15. This concept is based on targeting the largely interconnected cellular and molecular mechanisms that underlie ageing, collectively termed the ‘Hallmarks of Ageing’ [16]. The strength of this hypothesis lies in the wealth of scientific understanding of the biology of ageing and the remarkable progress that has been made recently in preclinical and human studies to identify interventions that prevent and reduce the burden of chronic disease with advancing age [17]. Undoubtedly, such an approach also has the potential to revolutionise the way in which we develop new drugs for multimorbidity by targeting the generic drivers rather than focusing on single diseases within a cluster.
The ageing process underpins the progressive loss of function, homeostasis, and resilience in multiple organ systems with time and, once a particular threshold of impairment is reached, leads to the development and accumulation of clinically discernible diseases and/or frailty [18]. Although most older adults with frailty also have multimorbidity, many people with multimorbidity are not phenotypically frail; that is, those at high risk of adverse outcomes, such as falls, disability, hospitalisation, and death, because of a loss of physiological reserve [18]. Despite being separate concepts, there is large overlap between frailty and multimorbidity, and frailty can usefully be considered as a tool to identify older people with multimorbidity who are particularly vulnerable to dramatic changes in their physical and mental wellbeing. Established models of frailty, such as the ‘Phenotype’ model developed by Fried [19] and the Rockwood ‘Cumulative Deficit’ model [20], often include aspects of function, such as mobility impairment or difficulty with daily living activities, which are commonly missing from multimorbidity models. Functional improvements form a core component of many evidence-based interventions to improve outcomes in frail individuals, and so measures of frailty can have an important role in geroscience-guided clinical trials targeting multimorbidity. The exact timing of multimorbidity onset is dependent on a multitude of genetic and environmental factors including, but not limited to, gender, ethnicity, obesity, lower socioeconomic status, and education level 8, 9, 10. Delaying the progression to multimorbidity from the first onset of disease is one possible approach to clinical trial design to test the geroscience hypothesis that is being piloted in the Targeting Aging with Metformin (TAME) trial (discussed in more detail later) [21]. Thus, the ageing hallmarks could represent novel therapeutic targets in age-related diseases that commonly cluster together in individuals with multimorbidity.
Hallmarks of ageing: potential targets for drug interventions in multimorbidity
It is now widely recognised that nine interdependent cellular and molecular mechanisms underpin the ageing process (the ‘Hallmarks of Ageing’) [16]. This biogerontological understanding of ageing has determined that the initiating process is the accumulation of damage to DNA and proteins, epigenetic alterations, and reduced genome stability. This results in cellular responses that produce the aged phenotype, including reduced nutrient sensing, altered cell communication, reduced stem cell turnover, mitochondrial dysfunction, reduced proteostasis, and, ultimately, cell senescence [16].
The integrity and stability of the genome is continuously challenged by exogenous agents [e.g., ultraviolet (UV) radiation and chemicals] as well as endogenous threats [e.g., replication-induced DNA errors and telomere shortening, spontaneous hydrolytic reactions, and reactive oxygen species (ROS)] [22]. Although organisms have developed DNA repair mechanisms to minimise these lesions [e.g., DNA damage responses (DDRs)] and maintain the length and functionality of chromosomes (e.g., telomeres), excessive DNA damage and insufficient DNA repair inevitably result in genomic instability with chronological age [22]. Epigenetic alterations, such as DNA methylation, histone modifications (e.g., methylation and acetylation), and chromatin remodelling, also take place constantly throughout the lifespan of an organism to control gene transcription in response to environmental stimuli. Progressive deregulation of the ‘the writers, readers, and erasers’ of epigenetic marks [e.g., DNA methyltransferases, histone acetyltransferases, methyl CpG binding proteins (MBPs), and histone demethylases] with age leads to epigenetic drift, which is characterised by a loss of the normal balance of regulatory landmarks within the epigenome 23, 24. The best example of this is in DNA methylation, where ageing causes a bidirectional shift in methylation patterns, resulting in global hypomethylation across the genome [25], as well as hypermethylation at key regions of genes (e.g., CpG islands) [26], which can eventually lead to chromatin compaction and gene silencing. Overall, these epigenetic alterations contribute to the attenuated responsiveness to internal and external signals, the loss of phenotypic plasticity, and reduced regenerative capacity of stem cells, as well as the rise of hyperproliferative somatic cells that promote cancer development [24]. These changes can also permit the reactivation of transposable elements (TEs), such as the retrotransposon LINE1, which commonly compromise genomic integrity [27].
Another mechanism that results in the accumulation of damage and initiation of the ageing process is the loss of proteostasis. The two main proteolytic systems involved in maintaining protein quality control, namely the ubiquitin-proteasome and the autophagy-lysosomal system, both become impaired with age [28]. Bulk lysosomal degradation, known as autophagy, is responsible for clearing long-lived proteins and damaged organelles, and is under tight control from multiple transduction pathways based on environmental signals [29].
An important regulator of autophagy is the mammalian target of rapamycin (mTOR) and, in its two multiprotein complexes mTORC1 and mTORC2, it forms part of an interconnected network of nutrient-sensing systems that affect a variety of cellular processes [29]. Whereas mTOR senses high amino acid concentrations and regulates almost all aspects of anabolic metabolism, the insulin-IGF-1 signalling (IIS) pathway responds to changing levels of glucose/insulin, as well as growth hormone (albeit indirectly via IGF-1) [29]. Thus far, multiple lines of evidence from longevity studies in animals [30] have suggested that strong anabolic and trophic signals, via mTOR and IIS, are major contributors to accelerated ageing, and the effects of dietary restriction (DR) on increasing lifespan in model organisms are likely mediated via modulation of these interconnected pathways. Other pertinent nutrient-sensing systems implicated in the ageing process involve AMPK and sirtuins, which detect changes in AMP and NAD + levels, respectively, to sense low energy states [29]. These nutrient sensors act in opposition to mTOR and IIS to signal nutrient scarcity and induce catabolic responses, and are accordingly upregulated with age [29].
Mitochondrial function is also perturbed with age as a result of mitochondrial DNA mutations, reduced mitochondriogenesis, destabilisation of the ATP-generating electron transport chain, and poorer quality control of damaged mitochondria by autophagy (known as mitophagy) [31]. Whereas mild mitochondrial dysfunction generates stress signals (e.g., ROS and low ATP production) that induce survival signals and trigger compensatory improvements in cellular fitness, in a process termed mitohormesis [32], with time these changes surpass a threshold that means oxidative stress becomes pathogenic [33].
Cellular senescence is a state of irreversible cell-cycle arrest, which can be induced by telomere shortening, DDRs, and mitochondrial stress [34]. Although senescent cells do not divide, they are metabolically active and secrete a variety of extracellular mediators, such as matrix metalloproteinases (MMPs), growth factors (vascular endothelial growth factor; VEGF), proinflammatory cytokines [e.g., interleukin-1 alpha (IL-1α) and IL-6), and chemokines (IL-8), collectively referred to as the senescence-associated secretory phenotype (SASP) [35]. The SASP has an important physiological role by recruiting immune cells to clear apoptotic cell debris for wound healing (e.g., by macrophages) and senescent cells themselves (e.g., by natural killer cells) [36]. However, with ageing, there is an accumulation of senescent cells, in part because of reduced immune-mediated elimination [37], thus causing aberrant tissue function and repair [34].
The SASP also contributes to the chronic, low-grade inflammation, termed ‘inflammageing’, which is a primary example of altered cell communication as a hallmark of ageing [38]. Inflammageing is characterised by reduced levels of anti-inflammatory cytokines, such as IL-10, and increased proinflammatory cytokines, such as IL-6 and IL-8, in the blood of older individuals [38]. Other potential contributors to inflammageing include impairment of the immune system with age (immunosenescence), increased adiposity, reduced physical activity, and leakage of microbial components from mucosal cavities into the blood because of reduced mucosal barrier integrity [39]. Importantly, the degree of inflammageing is associated with an increased risk of a range of chronic conditions, including cardiovascular disease, sarcopenia, osteoporosis, and dementia [40], and, thus, is a key potential driver of multimorbidity.
These highly interconnected core ageing processes not only provide a potential route to understanding the complex process of ageing and age-related diseases, but also offer an exciting opportunity to treat the generic drivers of multimorbidity to extend human healthspan.
Experimental evidence for targeting hallmarks of ageing in age-related disease and multimorbidity
Cellular senescence
Reducing the burden of senescent cells within tissues is one potential method of exploiting an ageing hallmark in the context of individual age-related diseases or multimorbidity, and has recently shown promise as a geroscience-informed therapeutic approach in a range of age-related diseases. Seminal papers in 2011 and 2016 showed that transgenic mice that selectively deleted senescent cells as they arose during midlife and old age had delayed tumorigenesis and age-related deterioration of several organs, including the heart, kidneys, eyes, skeletal muscle, and fat tissue 41, 42. Several existing drugs are able to induce apoptosis in senescent cells, so-called ‘senolytics’, including dasatinib (ephrin receptor inhibitor), quercetin (PI3 kinase pathway inhibitor), and navitoclax (Bcl-2/Bcl-xL anti-apoptotic protein inhibitor) 43, 44, 45. Such compounds have now been tested in several preclinical disease models, showing improvements in cardiac [46] and vascular function [47], atherosclerosis [48], osteoporosis [46], osteoarthritis [49], and cognitive decline in tau-dependent neurodegenerative disease [50], while also attenuating frailty and prolonging overall healthspan and lifespan [51]. Encouragingly, the first human trials of senolytics, a combination of dasatinib and quercetin, have reported promising early results, which are discussed in more detail later 52, 53. Screening of compound libraries has also led to the discovery of novel senolytics, including selective Bcl-xL inhibitors [54], fisetin 54, 55, heat-shock protein 90 (HSP90) inhibitors [56], and cardiac glycosides 57, 58.
In an alternative approach, senostatic drugs, which target the secretory activity of senescent cells and in doing so reduce inflammageing, have also been tested. Selective inhibition of the JAK1/2 pathway was shown to have senostatic activity, reducing systemic inflammation, improving metabolic function and fat tissue homeostasis, and enhancing physical capacity in old, frail mice [59]. Small-molecule p38 inhibitors have also been effective in reversing T cell senescence and boosting vaccine efficacy in mice [60], and reducing inflammation and boosting cutaneous immunity to viral challenge in older humans [61].
Despite the evidence from animal models that the removal of senescent cells can have a positive impact upon ageing in several organ systems and disease states, senolytics need to be tested stringently in humans to validate these preclinical findings and minimise potential translational risks. Accordingly, there are several clinical trials underway using senolytics in age-related disease/frailty cohorts (Table 1) to evaluate the safety, feasibility, and efficacy of such interventions.
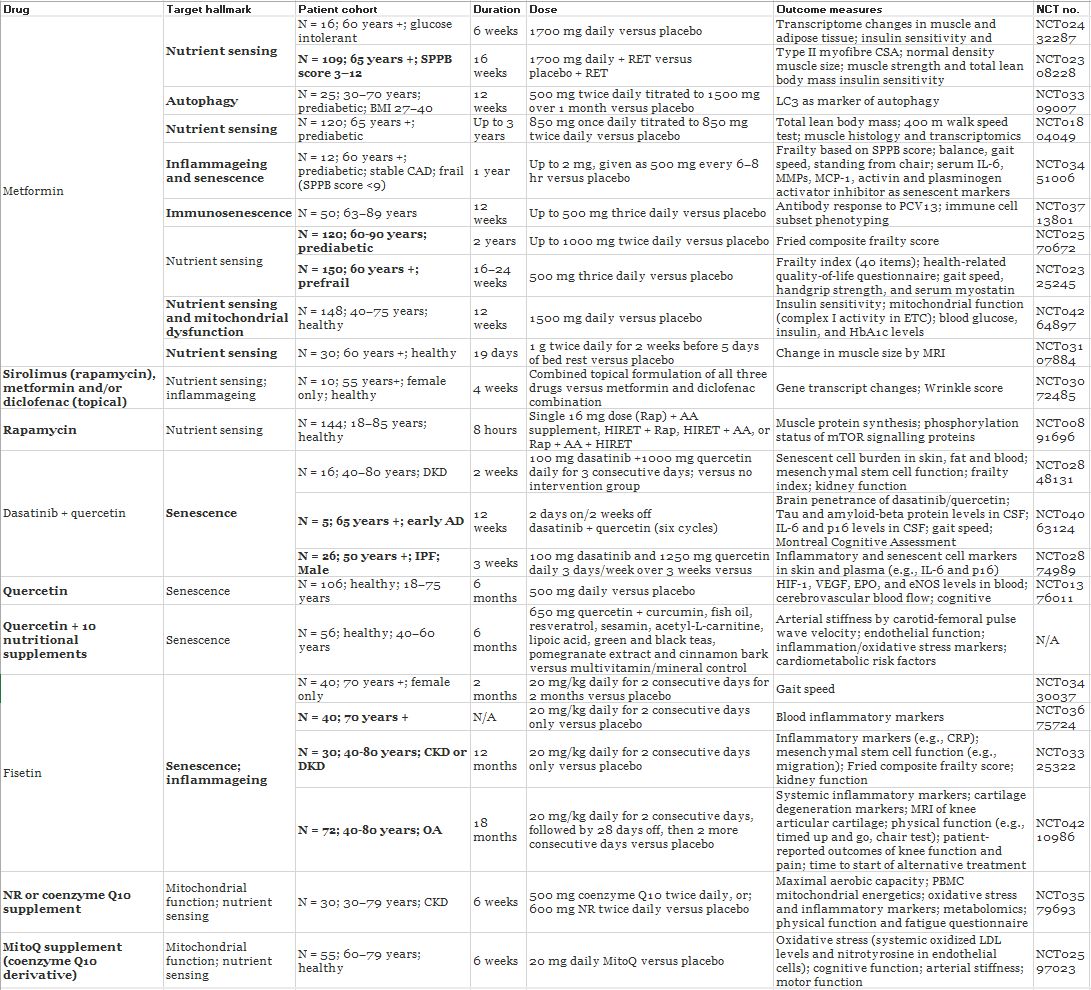
Table 1. Geroscience-guided clinical trials of drugs targeting hallmarks of ageing (a, b)
a
Given the wealth of preclinical evidence from animal models that ageing hallmarks can be targeted to delay and/or improve age-related diseases and frailty, human clinical trials are underway to investigate drugs known to target these core ageing processes. The use of diverse outcome measures that include biomarkers of ageing as well as more disease-specific measures will be vital to provide best possible evidence of efficacy for drug interventions.
b
Abbreviations: AA, amino acid; AAV-hTERT, adenovirus-associated human telomerase reverse transcriptase; AD, Alzheimer’s disease; BMI, body mass index; CAD, coronary artery disease; CFS, clinical frailty score; CKD, chronic kidney disease; CRP, C-reactive protein; CSA, cross-sectional area; CSF, cerebrospinal fluid; DKD, diabetic kidney disease; eNOS, endothelial nitric oxide synthase; EPO, erythropoietin; ETC, electron transport chain; HbAc1, glycated haemoglobin; HIF-1, hypoxia inducible factor; HIRET, high-intensity resistance exercise training; IL-6, interleukin-6; IPF, Idiopathic pulmonary fibrosis; LC3, microtubule-associated protein 1A/1B-light chain 3; LDL, low-density lipoprotein; MCP-1, monocyte chemoattractant protein-1; MMPs, matrix metalloproteinases; MRI, magnetic resonance imaging; NR, nicotinamide riboside; OA, osteoarthritis; PBMC, peripheral blood mononuclear cells; PCV13, pneumococcal conjugate vaccine; RET, resistance exercise training; SPPB, short physical performance battery; VEGF, vascular endothelial growth factor.
Mitochondrial dysfunction
Targeting the ageing hallmark of mitochondrial dysfunction has shown potential in several age-related pathologies. NAD levels decline with age and, although direct supplementation of NAD itself has had little success, NAD precursors, such as nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), have been more successful in increasing NAD tissue bioavailability [62]. For example, long-term NMN administration mitigates physiological decline in mice during normal ageing, with improvements in insulin sensitivity, plasma lipid profile, and eye and bone function, while simultaneously enhancing mitochondrial oxidative metabolism and attenuating age-associated mitonuclear protein imbalance in skeletal muscle [63]. In the context of obesity, NR supplementation protects against high-fat diet-induced metabolic abnormalities and defective mitochondrial function via the activation of SIRT1 and SIRT3 [64].
To date, the efficacy of NAD restoration in humans is less clear. Short-term NR supplementation has been shown to be well tolerated in middle-aged and older adults, leading to effective stimulation of NAD metabolism, as well as improvements in cardiovascular health [65] and decreased levels of circulating proinflammatory cytokines [66]. However, another clinical trial reported that NR supplementation in middle-aged, obese men with insulin resistance had no effect on NAD levels, respiratory capacity of skeletal muscle mitochondria, mitochondrial protein abundance, and mitochondrial fractional area [67]. Clinical trials using NR or Coenzyme Q10 derivatives are ongoing and will be vital in understanding the best possible way to target mitochondrial dysfunction and how this might impact other interconnected ageing hallmarks (Table 1).
Mitophagy and autophagy inducers
Urolithin A (UA), which is a natural dietary microflora-derived metabolite, specifically activates mitophagy and improved muscle health in animal models of ageing [68]. In a recent first-in-human clinical trial, synthetic UA supplementation had a good safety profile in old individuals and induced skeletal muscle gene expression changes indicative of improved mitochondrial function [69]. Spermidine, another inducer of mitophagy and autophagic flux in general, has also been tested as a dietary supplement in several preclinical animal models. So far, it has been shown to reduce hyperlipidaemia-induced atherosclerosis [70], protect against physiological cardiac ageing and heart failure [71], and reverse B cell senescence [72]. Spermidine-based nutritional interventions have now entered into a Phase IIb human clinical trial (SmartAge trial: NCT03094546) to investigate their effect on memory performance in older individuals with subjective cognitive decline and potential neurophysiological mechanisms of action [73].
Nutrient-sensing modulators
In addition to autophagy, other upstream components of the nutrient-sensing pathways can be manipulated to affect the ageing process. Although originally approved for its immunosuppressive effects in acute renal allograft rejection, rapamycin (sirolimus), which inhibits the mTOR complex that is central to nutrient and growth factor-sensing pathways, is arguably the most well-established therapeutic approach based on geroscience to date [74]. Although only mTORC1 is directly inhibited by rapamycin, chronic exposure inhibits mTORC2 assembly and is thought to be responsible for the adverse effects associated with long-term rapamycin use [75].
Although rapamycin is still being tested in clinical trials (Table 1), more specific mTORC1 inhibitors will be key in overcoming this problem and novel rapamycin analogues (also termed ‘rapalogs’) have already been shown to have high selectivity [76]. Thus far, acute treatment with rapamycin (or rapamycin analogues) to inhibit mTORC1 in animal models has been effective in delaying, or even reversing, almost every age-related disease or decline in function, including cancer, cardiovascular disease, kidney disease, obesity, neurodegenerative diseases, sarcopenia, and immune senescence [77]. To date, only four human studies have investigated mTORC1 inhibition in the context of ageing, mainly immune system decline. The first study reported that weekly treatment with the rapamycin analogue RAD001 (everolimus) for 6 weeks in healthy old individuals was shown to be relatively well tolerated and improved the immune response to influenza vaccination [78]. Similarly, 6-week low-dose combination treatment with RAD001 alongside the catalytic site mTOR inhibitor BEZ235 was safe and led to decreased infection rates over a 12-month period in old subjects owing, at least in part, to an improved response to influenza vaccination [79]. Although 8-week treatment with rapamycin at a higher dose of 1 mg per day was shown to be safe in healthy older individuals, no beneficial effects on cognitive and physical performance, or clinical laboratory and self-perceived health status, were observed [80]. Most recently, topical low-dose application of rapamycin to the skin in a group of over 40-year olds was shown to increase collagen VII levels and reduce senescent cell-associated p16-INK4A expression within the skin [81].
Metformin, which is approved for treatment of T2DM, targets several ageing hallmarks because it leads to decreased insulin levels, decreased IGF-1 signalling, inhibition of mTOR, inhibition of mitochondrial complex I in the electron transport chain, reduced endogenous ROS production, and activation of AMPK [82]. Metformin also favourably affects metabolic and cellular processes that contribute to inflammation, autophagy, and cellular senescence, although it is unclear whether it affects all these pathways directly, or whether the observed effects are downstream of its single action on one mechanism of ageing [82]. Beyond these cellular processes, there is growing evidence from preclinical studies that metformin can delay ageing and prolong lifespan and healthspan. In mice, for example, metformin increased lifespan by 4–6% and improved multiple healthspan indices, such as physical performance and insulin sensitivity. As expected, these improvements were accompanied by increased AMPK activity, decreased chronic inflammation, and reduced oxidative damage [83]. However, these preclinical findings must be interpreted with some caution for several reasons. First, although mice are genetically tractable and allow testing of how candidate drugs impact evolutionarily conserved mechanisms of ageing, they are by no means ideal as models of human ageing given the significant difference in lifespan compared with humans. Second, the lifespan-extending effects of metformin have not been replicated by the National Institute on Aging Interventions Testing Program, which tests the effects of drugs on longevity in three different locations using genetically heterogeneous mice. This might reflect the original effect being seen in only in male inbred mice, which are susceptible to obesity [84]. Third, high-dose metformin (1% w/w in diet) shortened the average lifespan in mice [83] and, thus, it will be important to identify safe dosing regimens in humans when determining the translational potential of metformin.
In humans, metformin has been shown to be effective in preventing T2DM in at-risk (obese or glucose-impaired) middle- and old-aged adults, while concomitantly improving cardiovascular disease risk factors and subclinical atherosclerosis [85]. In a pilot study in over 60-year olds with impaired glucose tolerance, 6 weeks of metformin use significantly altered metabolic and non-metabolic genes in both muscle and adipose tissue. Interestingly, these tissue-specific changes were predicted to be a result of metformin influencing upstream transcriptional regulators, such as mTORC1, as well as key ageing pathways linked to DNA repair, mitochondrial fatty acid oxidation, and inflammation [86]. Furthermore, metformin use has been associated with decreased cancer incidence, reduced cognitive impairment, lower rates of neurodegenerative diseases in older patients with T2DM, and reduced overall mortality in patients with T2DM as well as nondiabetics [82]. In contrast to these positive indications, metformin exposure in older patients with T2DM was associated with an increased risk of Alzheimer’s disease, vascular dementia, and Parkinson’s disease, and these observations were linked to increased dosage and duration of use [87]. A link between metformin use, increased risk of vitamin B12 and vitamin B6 deficiency, and cognitive dysfunction in older adults has also been reported [88]. Findings from recent clinical trials examining the effects of metformin alongside aerobic and/or resistance exercise training (AET/RET) in older adults were again contrary to expectation 89, 90. Metformin attenuated the improvement in insulin sensitivity and cardiorespiratory fitness after AET by inhibiting mitochondrial respiration in skeletal muscle [89], while preliminary results from the MASTERS trial (NCT02308228) found that long-term metformin use blunted the hypertrophic response to exercise and could not boost numbers of muscle-resident M2 (anti-inflammatory) macrophages after RET [90].
Such conflicting findings regarding metformin use in humans warrant future large-scale clinical trials. Currently, there are 11 clinical trials in the USA using metformin in patients with age-related conditions or frailty, aiming to reduce disease indicators or improve physical function respectively (Table 1). These trials will also be vital in discerning whether metformin can impact the root causes of ageing, beyond its known beneficial effects on metabolism. Monitoring effects on several hallmarks of ageing in these trials will have an important role in addressing this question.
Biomarkers of ageing for geroscience-guided clinical trials of multimorbidity
Any progress in preventing multimorbidity and late-life poor health will inevitably come from better predictors of their occurrence and the ability to intervene and monitor therapeutic responses in individuals at greatest risk of developing multimorbidity. The use of surrogate biological measures, biomarkers, to detect individual variability in the progress of ageing as a risk indicator (i.e., accelerated ageing) and to monitor responses to interventions will be vital in testing the geroscience hypothesis. According to the American Federation for Aging Research (AFAR), these biomarkers should ideally: (i) predict the rate of ageing (i.e., mark exactly where an individual is in their lifespan) and mortality independent of chronological age; (ii) monitor a fundamental ageing process and not the effects of disease; and (iii) allow for practical (i.e., via blood test or imaging) longitudinal tracking in laboratory animals and humans [91]. Although there is no broad consensus on biomarkers of ageing for use in clinical trials, an expert panel convened in 2017 proposed a list for use in geroscience-informed trials, which included markers for: inflammation [C-reactive protein (CRP) and IL-6); mitochondrial stress (GDF15); nutrient signalling (IGF-1 and fasting insulin); kidney function (cytostatin C); cardiovascular health (NT-proBNP); and metabolic ageing (glycated HbA1c) [21].
Several biomarkers of physiological state, including standardised measures of physical and mental capacity (e.g., cardiovascular and lung function, locomotion, strength, balance, and cognition) and systemic regulation of metabolism and immunity (e.g., insulin, IL-6, and CRP) have also been proposed for use in such trials [91]. Composite indices, such as the ‘Biological Age’ and ‘Pace of Aging’ scores, comprising many of these single biomarkers, can identify early-/middle-aged individuals at higher risk of impending multimorbidity because of accelerated ageing [92]. Other algorithmic measures of physiological vulnerability include the well-characterised ‘frailty phenotype’ [19], which is commonly used in the clinic, as well as the more recently developed frailty index (FI)-LAB [93], which uses commonly employed laboratory tests to generate a composite index of frailty. Such outcome measures might be more appropriate to monitor the effectiveness of interventions in older patients with established multimorbidity.
Recently, other indicators of biological age based on the epigenome [94], metabolome [95], and transcriptome [96] have been developed. For example, the ‘Horvath epigenetic clock’ is a multi-tissue DNA methylation-based predictor of chronological age based on the cumulative effect of the epigenetic maintenance system [94]. Deviation from the predicted chronological age of a subject using this tool is suggested as an indicator of their true biological age. This tool might have particular use in multimorbidity because it can elucidate tissue-specific accelerated ageing resulting from different diseases, and might enable us to identify which multimorbidity clusters should be targeted therapeutically. To date, these tools have not been extensively tested in clinical trials of drugs targeting ageing processes, but encouragingly they are starting to be adopted in some more recent geroscience-guided clinical trials, which are discussed in more detail in the following section.
Exemplars of geroscience-guided clinical trials
The exclusion of patients with multimorbidity from clinical trials is common, and often based on the proposition that the presence of comorbidities might dilute any potential benefits for the primary condition, or exacerbate any adverse effects of the treatment under investigation. The assessment of ongoing randomised controlled trials (RCTs) for patients with ten common chronic conditions registered between 2014 and 2015 in the UK found that 79% of these RCTs excluded patients with a chronic comorbidity, while only 12% of RCTs were investigating interventions specifically for patients with more than two chronic conditions [97]. A study of more recent industry-sponsored clinical trials of novel drug therapies for chronic conditions found that the mean comorbidity count for trial participants in the UK was approximately half that seen in community-based patients, with no comorbidities being found to be particularly well represented across any of the trials [98]. It is vital that any large-scale geroscience-guided clinical trials are designed to include individuals across the multimorbidity spectrum, including at-risk individuals, patients with a single disease, and patients with established multimorbidity, to determine how interventions can stop or slow progression from single disease to multimorbidity. Outcome measures should also reflect this transition by measuring time to next disease diagnosis or new disease incidence in a particular timeframe, and although expensive, will require multiple follow-ups to track longitudinal changes over months and potentially years. Despite the paucity of clinical trials that specifically target multimorbid patients, here we highlight exemplars of geroscience-guided clinical trials that have begun to pave the way for improved drug discovery in the prevention and treatment of multimorbidity.
.../...
.











