Hey everyone, Im making this post because think that the oxidative stress hypothesis of alzheimer's is frequently overlooked in favor of amyloid and tau.
I primarily looked into this to improve my nootropic stack. I'm not an expert, doctor or your mother and I'm certainly not the first to come up up with this, feedback is welcome.
I'm going to start off with the TLDR and some things i found promising: I believe that Alzheimer's is primarily the result of sirt6/glutathione deficiency which results the brain using tau to control double strand breaks without having enough resources (perhaps soluble ab42, more on that later) to effectively remove it.
So nac + glycine (glutathione precursors, better then serine imo) + carnitine + niacinamide riboside similar to the study below would likely be helpful
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9879258/
Nac with cofactors serine, niacinamide and l-carnitine improve ADAS-Cog scores by 29% (although i think the doses are a bit much)
In addition to these i think that ergothionine would further help to normalize this via the nrf2/ARE pathway.
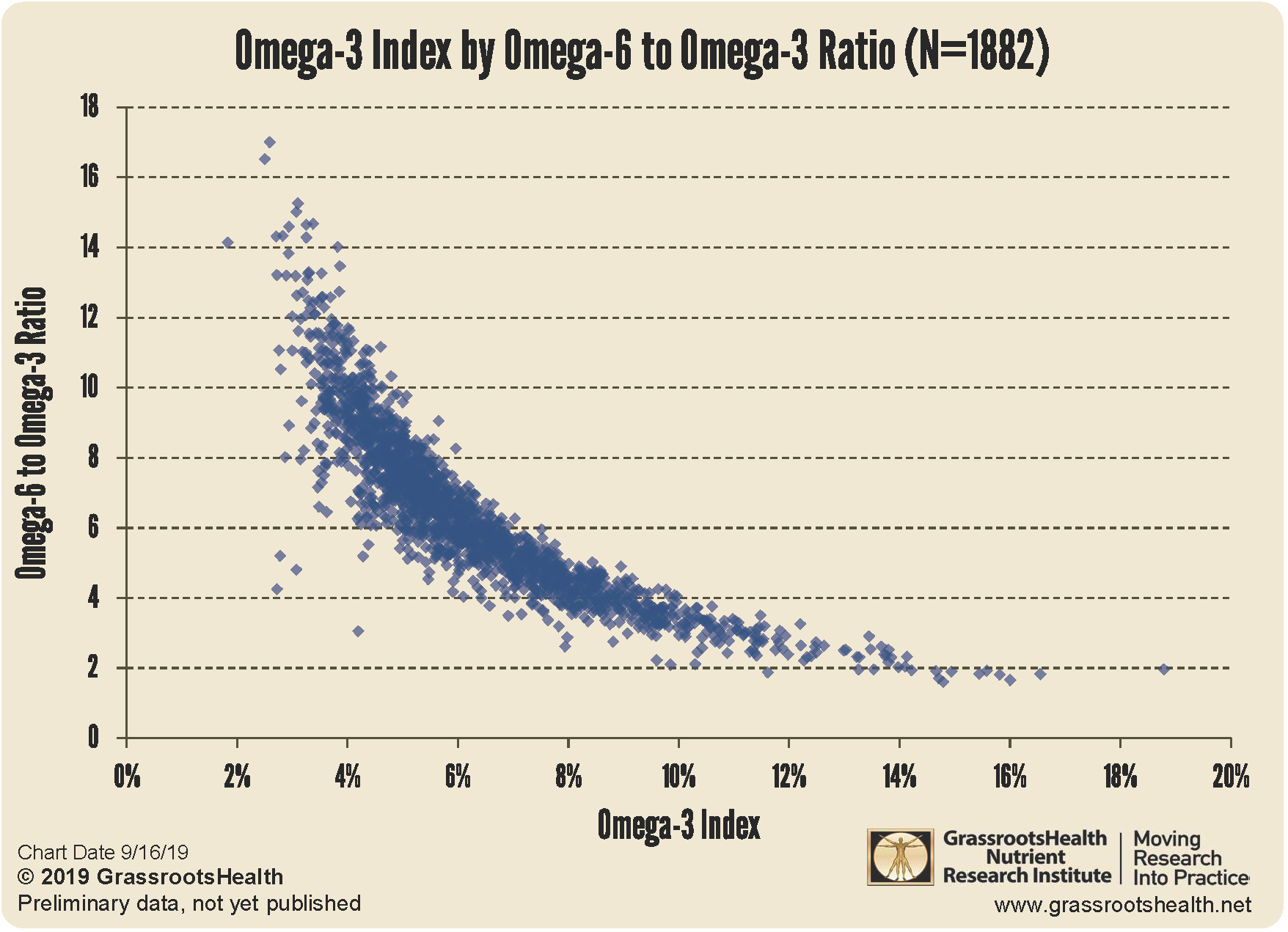
vitamin b12 + omega 3 are possibly synergistic in preventing brain atrophy
https://www.scienced...002916523277655
The addition of choline (or citicoline for uridine conversion) may improve synaptogenesis. In essence this part is basically the mrhappy stack.
https://pmc.ncbi.nlm...les/PMC4011061/
A valter longo style 3-5 day fasting mimicking diet is also likely to have a positive effect. It would clear out more trash while helping with neural stem cells.
https://www.ncbi.nlm...les/PMC9648488/
I also believe that vitamin k2 would help, for more then a few reasons.
Now onto the meat and potatoes, sorry in advance for the terrible formatting.
Glutathione and metal disruption
1. Glutathione is required for proper copper metabolism and a deficiency may cause a copper deficiency in the brain as well as a build of iron due to a lack of functional ceruloplasmin. You would expect to see a intracellular copper deficiency, a buildup of extracellular copper and dysfunction iron deposition due to a lack of ceruloplasmin,
https://pubmed.ncbi....h.gov/23426973/
“depleting cellular GSH using L-buthionine-sulfoximine (BSO) caused a 50% decrease in the initial rate of (64)Cu entry in HEK293 cells and other cell types.”
https://pubmed.ncbi....h.gov/28654115/
“copper levels were substantively decreased in all AD-brain regions, to 52.8-70.2% of corresponding control values, consistent with pan-cerebral copper deficiency”
2. Ferroptosis and iron deposition in the brain tracks strongly with Alzheimer’s and is activated via glutathione deficiency
https://www.ncbi.nlm...les/PMC9445173/
“Glutathione was found to be significantly depleted in mild cognitive impaired (P < 0.05) and Alzheimer’s disease patients (P < 0.001) as compared with healthy old participants. A significant higher
level of iron was observed in left hippocampus region for Alzheimer’s disease patients as compared with healthy old (P < 0.05) and mild cognitive impairment (P < 0.05).”
https://www.nature.c...598-022-22761-5
“The data clearly show that aggregated Aβ1-42 alone is significantly less toxic to hippocampal cells. Aggregated Aβ damages neurons, and glial cells proliferate to remove Aβ from the hippocampus. External prooxidant agents (Fe2+) or inhibition of internal antioxidant defence by BSO has more toxic effects on hippocampal cells than aggregated Aβ alone. Moreover, hippocampal cells fight against Aβ-induced damage more effectively than against oxidative damage.”
https://stemcellres....287-024-03644-0
“Our study confirms that ferroptosis participates in regulating fat graft survival and that GSH exerts a protective effect by inhibiting ferroptosis.”
3. One of the most important oxidants involved in the progression of Alzheimer’s appears to be superoxide. This is primarily quenched by superoxide dismutase which requires copper to function. It isn’t a large jump to suppose that the increased superoxide is in part a result of reduced sod (due to copper deficiency) and glutathione.
https://www.ncbi.nlm...les/PMC9648488/
“These results indicate that FMD cycles delay cognitive decline in AD models in part by reducing neuroinflammation and/or superoxide production in the brain.”
https://pubmed.ncbi....h.gov/19666610/
“We found that overexpression of SOD-2 decreased hippocampal superoxide, prevented AD-related learning and memory deficits, and reduced Abeta plaques. Interestingly, SOD-2 overexpression did not affect the absolute levels of Abeta(1-40) and Abeta(1-42), but did significantly reduce the Abeta(1-42) to Abeta(1-40) ratio, thereby shifting the balance toward a less amyloidogenic Abeta composition”
Glutathione and mitophagy
1. One of the primary differences between healthy people with amyloid and people with Alzheimer’s is in Alzheimer’s mitophagy is impaired. Glynac largely restores this.
“Only GlyNAC supplementation in OA significantly improved mitophagy in OAG-16w, together with a nonsignificant improvement in autophagy (LC3AB expression”
https://academic.oup...78/1/75/6668639
2. Glutathione deficiency in treg cells causes mistakes to build up as their mitophagy is impaired. It isn’t well explored but it is possible that treg cells work progressively worse as the disease progresses
“Gclc-deficient Tregs show increased serine metabolism, mTOR activation and proliferation but downregulated FoxP3. Limitation of cellular serine in vitro and in vivo restores FoxP3 expression and suppressive capacity to Gclc-deficient Tregs. Our work reveals an unexpected role for GSH in restricting serine availability to preserve Treg functionality.”
https://www.ncbi.nlm...les/PMC7265172/
“Overall, our results indicate an impaired mitophagy in Tregs during an autoimmune response that results in accumulation of dysfunctional mitochondria that might contribute to loss of Treg suppressive function.”
https://ard.bmj.com/...6/Suppl_1/A36.3
3. Treg cells deficient in glutathione upregulate serine metabolism, similarities in serine can also be seen in the Alzheimer’s brain. Such as the increased PHGDH enzyme which is used to produce serine
“In analyzing brain tissue, researchers observed a trend consistent with their previous findings in blood samples: expression levels of the gene coding for PHGDH were consistently higher in adults with different stages of Alzheimer’s disease, even the early stages before cognitive symptoms manifested.” https://today.ucsd.e...ing-supplements
Glutathione and amyloid/tau
1. Oxidative stress and lack of glutathione likely happens first
https://pubmed.ncbi....h.gov/36278355/
“The order of pathologic progression in the 3xTg-AD mouse was loss of GSH (oxidative redox shift) followed by a pAkt/tAkt metabolic shift in CA1, iAβ accumulation in CA1, and extracellular Aβ deposition. Upstream targets may prove strategically more effective for therapy before irreversible changes.”
2. Tau is likely used as a stopgap during sirt6 deficiency in response to dna damage.
https://pubmed.ncbi....h.gov/33910019/
“Here, we show that Tau hyper-acetylation at residue 174 increases its own nuclear presence and is the result of DNA damage signalling or the lack of SIRT6, both causative of neurodegeneration.”
https://www.nature.c...003-022-03312-0
In vitro studies using primary mouse cortical neurons show that non-p-tau accumulates perinuclearly together with the tubulin after DSB induction with etoposide, followed by the accumulation of phosphorylated tau. Moreover, the knockdown of endogenous tau exacerbates DSB in neurons, suggesting the protective role of tau on DNA repair.
https://www.nature.c...419-022-05542-w
Our analysis reveals that SIRT6 is a central regulator of mitochondrial activity in the brain. SIRT6 deficiency in the brain leads to mitochondrial deficiency with a global downregulation of mitochondria-related genes and pronounced changes in metabolite content
https://www.research...acetylase_SIRT6
“Moreover, the brains of these mice show increased signs of DNA damage, cell death, and hyperphosphorylated Tau—a critical mark in several neurodegenerative diseases. Mechanistically, SIRT6 regulates Tau protein stability and phosphorylation through increased activation of the kinase GSK3a/”
3. Sirt6 is preserved by p53 which is preserved by glutathione. Ros appears to inhibit p53 functioning. Upregulation of p53 by nutlin-3 (a mdm2 inhibitor, like curcumin, genistein) prevents sirt-6 reduction induced by ab42
https://www.nature.c...598-023-35533-6
GAS-STING-interferon signalling was impaired in AD and was accompanied by a depletion of STING protein from Golgi and a failure to elevate interferon despite the presence of DSBs. The results suggest that oxidation of p53 by ROS could inhibit the DDR and decrease its ability to orchestrate DSB repair by altering the oligomerization state of p53.”
https://www.pnas.org...pnas.1718819115
Our findings suggest a pivotal role for cellular NAD+ depletion upstream of neuroinflammation, pTau, DNA damage, synaptic dysfunction, and neuronal degeneration in AD. Interventions that bolster neuronal NAD+ levels therefore have therapeutic potential for AD.
4. There is evidence that soluble ab42 may be protective as well as help to dissolve tau.
https://www.scienced...40911112040.htm
https://pubmed.ncbi....h.gov/36693165/
“we discovered that fresh Aβ42 could protect Tau against the formation of disulfide cross-linked dimers. We showed that the monomeric and small Aβ oligomers (the "nonamyloidogenic Aβ") efficiently disassembled tau dimers and heparin-induced Tau oligomers to recover Tau monomers. Interestingly, Aβ serves the role of an antioxidant to prevent disulfide bond formation”
https://pubmed.ncbi....h.gov/39259179/
“Higher CSF Aβ42 levels after exposure to anti-Aβ drugs are independently associated with slowing cognitive impairment and clinical decline. Increases in Aβ42 may represent a mechanism of potential benefit of anti-Aβ monoclonal antibodies in AD”
5. If tau is removed via a treatment and then comes back, this is likely due to the fact that glutathione deficiency still exists and is unable to repair the damage left by the tau.
Glutathine in amaloid involving conditions/miscellanious
1. Nac with cofactors serine, niacinamide and l-carnitine improve Alzheimer scores by 29% https://www.ncbi.nlm...les/PMC9879258/
2. Nac largely reduces amyloid in a rare amyloid skin condition
https://www.nature.c...467-021-22120-4
“Incubations of either lysate or supernatant containing L68Q-hCC with reducing agents glutathione or N-acetyl-cysteine (NAC) breaks oligomers into monomers. Six L68Q-hCC carriers taking NAC had skin biopsies obtained to determine if hCC deposits were reduced following NAC treatment. Remarkably, ~50–90% reduction of L68Q-hCC staining was observed in five of the treated carriers suggesting that L68Q-hCC is a clinical target for reducing agents.”
3. Glutathione deficiency increases the ab40/42 ratio in mice https://febs.onlinel...1873-3468.14895
4. N-Acetylcysteine Amide against Aβ-Induced Alzheimer’s-like Pathology in Rats https://www.ncbi.nlm...es/PMC10454451/