The early free radical theory of aging was based on Dr. Denham Harman's remarkable insight that much of the cellular and molecular damage of aging bore strong resemblance to damage he observed in organisms exposed to ionizing radiation.(1) In subsequent decades, the theory -- and its later refinement into the mitochondrial free radical theory of aging(2,3) -- has gained wide acceptance, even as the challenges against it have mounted and risen in sophistication. Despite the clear evidence that certain sources of reactive oxygen species (ROS*), and certain kinds of oxidative stress and damage, contribute to the degenerative aging process,(4,5) it has become equally clear that the early, and still widely-invoked, oversimplification that all free radicals of any sort, in any context, are deleterious, cannot be sustained.
Perhaps the most profound exception to the rule of the inherent toxicity of ROS can be laid at the feet of the engines of evolution. Over evolutionary time, natural selection has "learned" to harness these ubiquitous reactive small molecules for use in cellular signal transduction, playing initially-surprising but ultimately-comprehensible roles in such processes as insulin signaling and the adaptive response to resistance exercise.(6,24) A common mechanism of redox signaling is via chemical reactions of specific ROS (primarily singlet oxygen or hydrogen peroxide) with specific atoms (usually a cysteine residue) in transcription factors or other regulatory elements: the ensuing covalent modification of the protein alters its activity, providing a mechanism for the cell to respond dynamically to shifts in intracellular redox tone.

Figure 1. Bacterial examples of ROS regulation of transcription factors. Reproduced from (24).
Two recent studies have now provided especially salient examples of this novel form of the "oxygen paradox."
Antioxidants Impair Autophagy
David Rubinsztein's laboratory at the Cambridge Institute for Medical Research focuses on diseases of aggregate-prone proteins, and especially disorders caused by codon reiteration mutations, such as Huntington’s disease (HD). In the earlier study,(7) Dr. Ben Underwood and colleagues in Rubinsztein's lab followed up on earlier research documenting the role of ROS in mediating the upregulation of autophagy in response to starvation and a range of pharmacological agents, including the prominent case of rapamycin.
The importance of autophagy and oxidative stress to both pathology and potential therapies for neurodegeneration has led us to investigate their relationship further. A diverse range of stimuli that induce both ROS and autophagy have been described and autophagy induction by these agents is antagonized by antioxidants ... Here we show that not all autophagy inducers significantly increase ROS. However, many antioxidants inhibit both basal and induced autophagy ...When autophagy induction is mediated by an increase in ROS, increases in autophagy can be inhibited with ROS scavengers. We next tested whether the impairment of autophagy by antioxidants was limited to situations where the autophagy-inducing agent also significantly increased ROS. We tested the ability of a variety of thiol antioxidants (including those proposed for treatment of HD) to ameliorate the induction of autophagy by trehalose (which does not increase levels of ROS) in COS-7 cells.
We found that NAC N-acetylcysteine (NAC), cystamine (a drug proposed for use in HD ... which is also metabolized to the antioxidant L-cysteine (19)) and glutathione were all able to significantly ameliorate the induction of autophagy by trehalose in a dose-dependent fashion, as measured by levels of the autophagy marker, microtubule-associated protein 1 light chain 3 II (LC3-II) ... NAC [also] impaired the increase in LC3-II associated with rapamycin treatment and also decreased LC3-II compared with the basal state. We confirmed the effect of NAC on basal and inducible autophagy in human primary cortical neurons ... and in HeLa cells (Fig. 2F). Pre-treatment with cystamine had similar effects on basal autophagy ...
[W]e examined [several antioxidants'] effect on the mTOR [mammalian Target of Rapamycin] pathway, a classical negative regulator of autophagy, by looking for changes in the phosphorylation status of an mTOR kinase substrate 4E binding protein 1 (4E-BP1) and a protein dependent on mTOR substrate kinase activity, ribosomal protein S6 (S6). Vitamin E enhanced the activity of mTOR but, surprisingly, NAC appeared to be inhibiting mTOR activity—an effect that would be expected to increase, rather than decrease, autophagy. ...
Starvation-induced autophagy is associated with increased levels of ROS and has been shown to be mediated by the activation of c-Jun N-terminal protein kinase 1 (JNK1) ... NAC is able to inhibit both ROS accumulation and autophagy in starved cells. ... NAC and glutathione inhibited the activation of JNK and decreased the phosphorylation of Bcl-2. ...
In order to investigate the physiological relevance of our findings, we examined the effect of NAC on starvation-induced autophagy in mice. ... As expected, starvation strongly increased hepatic LC3-II levels, but this increase was significantly smaller in mice that had been pre-treated with NAC. ...
By blocking autophagy, antioxidant drugs can increase the levels of aggregate-prone proteins associated with neurodegenerative disease. In fly and zebrafish models of Huntington's disease, antioxidants exacerbate the disease phenotype and abrogate the rescue seen with autophagy-inducing agents.Thus, the potential benefits in neurodegenerative diseases of some classes of antioxidants may be compromised by their autophagy-blocking properties.(7)
From an evolutionary point of view, one can see a highly-intuitive hypothesis to explain of this phenomenon. When damaged biomolecules begin to accumulate in the cell, it will often lead to oxidative stress downstream; equally, conditions of high oxidative stress will predictably lead to an increase in the production of such damaged cellular constituents. This could create selective pressure favoring the retention of redox-sensitive protein sites on regulatory elements in the degradative machinery, allowing for upregulation of the proteasomal or (in this case) lysosomal machinery to clear the offending aggregates.
ROS-Sensitive Regulation of Stem Cell Proliferative Homeostasis
Perhaps more surprising is a more recent report from Heinrich Jasper's lab at the University of Rochester, showing that the antioxidant Cap'n'collar transcription factor Nrf2 -- which suppresses intracellular oxidative stress by inducing genes involved in the production of the cellular antioxidants glutathione and the thoredoxins, along with peroxiredoxin enzymes -- is constitutively active in Drosophila intestinal stem cells.(8) The high level of CncC (Drosophila Cap'n'collar group, including Nrf2) activity in intestinal stem cells substantially blunts the paraquat-induced rise in oxidative stress, while RNA interference (RNAi)-mediated repression of CncC translation leads to a rise in basal oxidative stress and elevated stem cell proliferation.
The role of ROS in regulating stem cell proliferation was also confirmed by other means. Thus, knockdown components of complex I of the mitochondrial electron transport chain via RNAi leads to a rise in intracellular ROS concentration, with a concomitant rise in stem cell proliferation. Contrariwise, overexpression the genes encoding the rate-limiting enzyme for glutathione biosynthesis, or of those encoding a thioredoxin peroxidase whose expression can be stimulated by CncC overexpression, simultaneously lowers ROS concentration and proliferation rate in these stem cells. Most dramatically, overexpression of these same genes also delays the onset of the age-associated increase in gut stem cell mitosis observed in wild-type flies.(8)
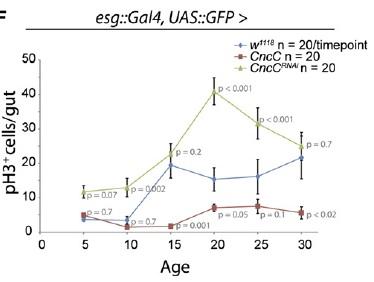
Figure 2. Elevating CncC Activity (and Nrf2) Delays Age-Related Rise in Uncontrolled Stem Cell Proliferation. Reproduced from (8).
Thus, Nrf2-induced suppression of ROS in these cells was shown to enforce quiescence. The counterintuitive corollary of this finding is that in order to respond to inflammation and oxidative stress with stem cell proliferation to ensure tissue renewal, the "protective" antioxidant-induction activity of Nrf2 must be repressed. Accordingly, induced coexpression of Nrf2/CncC was shown to inhibit the defensive proliferative response induced by a range of stimuli, including oxidative stress (paraquat), mitogenic signaling through the insulin receptor, expression of the stress-response protein JNK (which is normally activated by stressors such as inflammatory cytokines, heat or osmotic shock, ultraviolet irradiation, and bacterial enterotoxins), by exposure to bacterial lipopolysaccharide, or by hyperplasia induced by Notch-targeting RNAi.(8) The negative regulation required to rein in Nrf2 and allow stem cells to proliferate under stress conditions was shown to be provided by Keap1, which sequesters Nrf2 in the cytosol and ultimately ubiquitinates it for subsequent proteolytic degradation.(8)
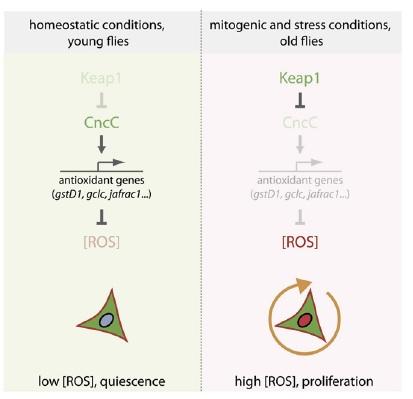
Figure 3. Nrf2, ROS, and the Regulation of Stem Cell Proliferation. Reproduced from (8).
These findings shed light on an earlier report from the same group:
In aging flies, the intestinal epithelium degenerates due to over-proliferation of intestinal stem cells (ISCs) and mis-differentiation of ISC daughter cells, resulting in intestinal dysplasia. Here we show that conditions that impair tissue renewal [ectopic activation of Notch, or overexpression of JNK or insulin signaling] lead to lifespan shortening, whereas genetic manipulations that improve proliferative homeostasis extend lifespan. These include reduced Insulin/IGF or Jun-N-terminal Kinase (JNK) signaling activities, as well as over-expression of stress-protective genes [the heat-shock protein Hsp68 or the peroxiredoxin Jafrac1] in somatic stem cell lineages. Interestingly, proliferative activity in aging intestinal epithelia correlates with longevity over a range of genotypes, with maximal lifespan when intestinal proliferation is reduced but not completely inhibited [my emphasis]. Our results highlight the importance of the balance between regenerative processes and strategies to prevent hyperproliferative disorders and demonstrate that promoting proliferative homeostasis in aging metazoans is a viable strategy to extend lifespan.(9)
Figure 4. Mild Repression of Stem Cell Proliferation Extends Lifespan in Drosophila. Reproduced from (9).
Implications for Intervention in Aging
Long prior to these recent findings, earlier research not discussed by these authors had shown that life-extending Calorie restriction (CR) in rodents exhibits this same property, placing cell replication under a more "thrifty" regulatory regime and thereby maintaining tisue-renewal capacity with age (16-19) and contributing to the CR animals' cancer resistance (20-23) and extended lifespan.(21) Established mediators of this more aggressive control of cell proliferation include reduced signaling through the insulin, insulin-like growth factor, and mammalian target of rapamycin (mTOR) signaling pathways. More recently, and apparently independently of all three lines of inquiry, still another group has reported that much of the cancer-protective effect of CR in mice is dependent on upregulation of Nrf2 activity.(10) Taken together, these findings intimate the possibility that the same Nrf2-dependent control of stem cell proliferation, dysplasia, and lifespan observed in Drosophila(8,9) may extend to the well-established rodent CR model of retarded aging.(10,16-23)
Interestingly, several dietary phytochemicals thought to have chemopreventive activity have also been shown to increase Nrf2 activity in vivo in at least some tissues after oral administration.(11-15) It is generally assumed that the mechanistic basis for the apparent role of Nrf2 induction in a cancer-preventive action of such agents and of CR is through the quenching of ROS and the detoxification of carcinogenic and mutagenic compounds.(10-15) This new research(8) suggests Nrf2-mediated inhibition of stem cell proliferation as an additional or alternative mechanism.
As Underwood et al also highlight for their own research into autophagy,(7) the new revelations about Nrf2 in regulation of stem cell proliferation(8-10) suggests the possibility that antioxidant supplementation may impede the protective effects of these interventions. Such pleiotropic effects could hypothetically have contributed to the multiple failures of antioxidant therapies to extend lifespan in experimental animals, or to reduce the incidence of cardiovascular disease and cancer in large clinical trials in humans. More speculatively, it may also raise some concerns about efforts to restore stem cell mobilization in aging tissues by elevating Notch signaling. And further complicating the interpretation of these findings or their use as the basis for interventions in age-related disease, mutations leading to constitutive activation of Nrf2 have been associated with several kinds of cancer -- most prominently of the lung -- in humans.(25)
As in previous cases we have highlighted, paradoxical findings and dilemmas such as these challenge the wisdom of efforts to retard the degenerative aging process by manipulating metabolic regulatory machinery and its dynamic intermediates. Homeostasis relies upon the ability of the organism to sense its internal and external environment, and respond to it adaptively; our best efforts to improve on the homeostatic regulatory machinery, or to selectively alter the dynamic environment itself, will inevitably tend to founder on the rocks of our ignorance of metabolism, and sink in the swirling seas of its complexity. The "engineering" heuristic of intervention in the degenerative aging process offers a means to traverse these perilous waters by figuratively flying above them, leaving the regulation of homeostasis to the finely-tuned machinery that has already been shaped by the complex calculus of deep evolutionary time -- machinery which has kept ourselves, our species, and our evolutionary ancestors alive for all of our lives. Rather, rejuvenation biotechnology will maintain youthful health and vigor through the direct repair, removal, replacement, and rendering-harmless of the damage itself instead of second-guessing its regulators.
Note
* I will here adhere to this conventional terminological convenience, with the understanding that the species under discussion include some molecules and ions that do not meet the literal meaning of the term, such as reactive nitrogen species and reduced transition metal ions.
References
1: Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956 Jul;11(3):298-300. PubMed PMID: 13332224.
2: Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972 Apr;20(4):145-7. PubMed PMID: 5016631.
3: de Grey ADNJ. The mitochondrial free radical theory of aging. Austin, TX: Landes Bioscience, 1999, 212pp, hardcover (ISBN 1-57059-564-X).
4: Pamplona R, Barja G. Highly resistant macromolecular components and low rate of generation of endogenous damage: two key traits of longevity. Ageing Res Rev. 2007 Oct;6(3):189-210. Epub 2007 Jul 13. Review. PMID: 17702671 [PubMed - indexed for MEDLINE]
5: Pamplona R. Mitochondrial DNA Damage and Animal Longevity: Insights from Comparative Studies. J Aging Res. 2011, Article ID 807108, 9 pages. doi:10.4061/2011/807108
6: Rudolph TK, Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci Signal. 2009 Sep 29;2(90):re7. Review. PubMed PMID: 19797270.
7: Underwood BR, Imarisio S, Fleming A, Rose C, Krishna G, Heard P, Quick M, Korolchuk VI, Renna M, Sarkar S, García-Arencibia M, O'Kane CJ, Murphy MP, Rubinsztein DC. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum Mol Genet. 2010 Sep 1;19(17):3413-29. Epub 2010 Jun 21. PubMed PMID: 20566712; PubMed Central PMCID: PMC2916709.
8: Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011 Feb 4;8(2):188-99. PubMed PMID: 21295275; PubMed Central PMCID: PMC3035938</p>Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011 Feb 4;8(2):188-99. PubMed PMID: 21295275; PubMed Central PMCID: PMC3035938
9. Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010 Oct 14;6(10):e1001159. PubMed PMID: 20976250; PubMed Central PMCID: PMC2954830.
10: Martín-Montalvo A, Villalba JM, Navas P, de Cabo R. NRF2, cancer and calorie restriction. Oncogene. 2011 Feb 3;30(5):505-20. Epub 2010 Nov 8. PubMed PMID: 21057541.
11: McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J Nutr. 2004 Dec;134(12 Suppl):3499S-3506S. PubMed PMID: 15570060.
12: Shen G, Xu C, Hu R, Jain MR, Gopalkrishnan A, Nair S, Huang MT, Chan JY, Kong AN. Modulation of nuclear factor E2-related factor 2-mediated gene expression in mice liver and small intestine by cancer chemopreventive agent curcumin. Mol Cancer Ther. 2006 Jan;5(1):39-51. PubMed PMID: 16432161.
13: Yuan JH, Li YQ, Yang XY. Inhibition of epigallocatechin gallate on orthotopic colon cancer by upregulating the Nrf2-UGT1A signal pathway in nude mice. Pharmacology. 2007;80(4):269-78. Epub 2007 Jul 26. PubMed PMID: 17657175.
14: Yuan JH, Li YQ, Yang XY. Protective effects of epigallocatechin gallate on colon preneoplastic lesions induced by 2-amino-3-methylimidazo[4,5-f ] quinoline in mice. Mol Med. 2008 Sep-Oct;14(9-10):590-8. PubMed PMID: 18596869; PubMed Central PMCID: PMC2442020.
15: Balstad TR, Carlsen H, Myhrstad MC, Kolberg M, Reiersen H, Gilen L, Ebihara K, Paur I, Blomhoff R. Coffee, broccoli and spices are strong inducers of electrophile response element-dependent transcription in vitro and in vivo - Studies in electrophile response element transgenic mice. Mol Nutr Food Res. 2011 Feb;55(2):185-97. doi: 10.1002/mnfr.201000204. Epub 2010 Sep 8. PubMed PMID: 20827676.
16: Chou MW, Shaddock JG, Kong J, Hart RW, Casciano DA. Effect of dietary restriction on partial hepatectomy-induced liver regeneration of aged F344 rats. Cancer Lett. 1995 May 8;91(2):191-7. PubMed PMID: 7767909.
17:Wolf NS, Penn PE, Jiang D, Fei RG, Pendergrass WR. Caloric restriction: conservation of in vivo cellular replicative capacity accompanies life-span extension in mice. Exp Cell Res. 1995 Apr;217(2):317-23. PubMed PMID: 7698231.
18: Holcomb VB, Keck VA, Barrett JC, Hong J, Libutti SK, Nunez NP. Obesity impairs wound healing in ovariectomized female mice. In Vivo. 2009 Jul-Aug;23(4):515-8. PubMed PMID: 19567384.
19: Reed MJ, Penn PE, Li Y, Birnbaum R, Vernon RB, Johnson TS, Pendergrass WR, Sage EH, Abrass IB, Wolf NS. Enhanced cell proliferation and biosynthesis mediate improved wound repair in refed, caloric-restricted mice. Mech Ageing Dev. 1996 Jul 31;89(1):21-43. PubMed PMID: 8819104.
20: Bursch W, Grasl-Kraupp B, Wastl U, Hufnagl K, Chabicovsky M, Taper H, Schulte-Hermann R. Role of apoptosis for mouse liver growth regulation and tumor promotion: comparative analysis of mice with high (C3H/He) and low (C57Bl/6J) cancer susceptibility. Toxicol Lett. 2004 Apr 1;149(1-3):25-35. Review. PubMed PMID: 15093245.
21: Koizumi A, Wada Y, Tuskada M, Kayo T, Naruse M, Horiuchi K, Mogi T, Yoshioka M, Sasaki M, Miyamaura Y, Abe T, Ohtomo K, Walford RL. A tumor preventive effect of dietary restriction is antagonized by a high housing temperature through deprivation of torpor. Mech Ageing Dev. 1996 Nov 29;92(1):67-82. PubMed PMID: 9032756.
22: Koizumi A, Tsukada M, Hirano S, Kamiyama S, Masuda H, Suzuki KT. Energy restriction that inhibits cellular proliferation by torpor can decrease susceptibility to spontaneous and asbestos-induced lung tumors in A/J mice. Lab Invest. 1993 Jun;68(6):728-39. PubMed PMID: 8515658.
23: Jin YH, Koizumi A. Decreased cellular proliferation by energy restriction is recovered by increasing housing temperature in rats. Mech Ageing Dev. 1994 Jul;75(1):59-67. PubMed PMID: 9128754.
24: D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007 Oct;8(10):813-24. Review. PubMed PMID: 17848967.
25: Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009 Apr;34(4):176-88. Epub 2009 Mar 25. Review. PubMed PMID: 19321346.
View the full article










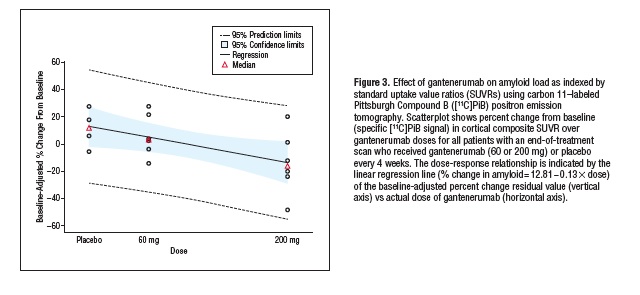
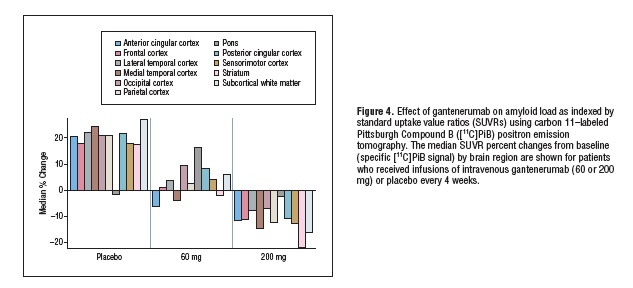
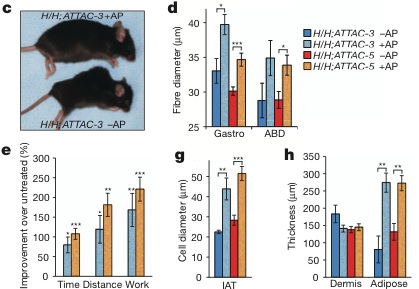 H/H;INK-ATTAC Mice" />
H/H;INK-ATTAC Mice" />