































By James P Watson with contributions and assistance by Vince Giuliano
This is Part 2 of what will likely be a three or four-part series of blog entries related to the metabolic cofactor NAD+. The Part 1 blog entry provided an overview treatment of the NAD World and its nuances: identified the major molecular entities involved, their roles, health and longevity ramifications, the reasons for the current excitement, and begin to clarify what is actually known and what the remaining uncertainties are. We also discussed a few important facets of the general topic which are ignored in many of the current publications, such as the high degree of circadian regulation of NAD+ .and certain proteins that regulate NAD+ and its cousins such as CD38 and CCAR2,
Introduction to part two by Vince Giuliano.
Personally, I am finding that developing a thorough understanding of the World of NAD+, including what is relevant about all of the Sirtuin’s, cofactors and pathways, is one of the more-difficult intellectual challenges I have ever faced. I am reminded of the process of understanding quantum physics I went through as a university student. And the difficulty of getting my mind around a complex of strange and completely non-intuitive concepts, dreaming strange dreams at night until I was finally willing to live with what I could not completely fathom. I am reminded of the wisdom of the often-quoted Winston Churchill graduation speech to his high school where he virtually flunked out years earlier “never give up. Never give up. Never give up! Never. Never. Never, (ref)” For personally I think the NAD World is extremely important pathway and I want to understand it to the limit of my capability and what is actually known. In that light, I acknowledge that this present blog entry covers much of the same territory as that already covered in the Part 1 blog entry. However, the coverage is from a different set of perspectives and includes much new material. While there is significant redundancy between what is discussed here and in the Part 1 blog entry, and additional redundancy within this blog entry itself, I believe this can contribute to understanding.
This blog entry concentrates on the reasons for focusing on NAD+, particularly with respect to interventions that are seriously likely to lead to longer healthier lives. It discusses molecular processes in the NAD salvage cycle that are responsible for the health-inducing and life-extending properties of calorie restriction and further discuss the key roles of Sirtuins, SIRT1, SIRT6 and SIRT7 in particular. It goes into more detail in several areas covered less- deeply in the Part 1 blog entry. It points to some difficult-to-integrate contrarian research and basic uncertainties. Also, Jim amfd I are centrally concerned with developing a set of biomarkers that can tell us what the various approaches to enhancing body levels of NAD+ actually accomplish or fail to accomplish. So many of our comments are addressed to this point.
We expect Part 3 will go into additional detail more explicitly describing a number of strategies currently being researched or proposed for maintaining high constituent levels of NAD+. We expect to further explain reasons why simply enhancing NAD+ levels might not work to increase health or longevity and speculates on what else might have to be done in addition. Finally, we expect further to discusses a variety of practical biomarkers to enable comparative evaluation of the NAD+ enhancement strategies, including information that might be provided by a new generation of health and fitness smart watches, smart bands, and other consumer measuring devices just now coming on the market.
Like many of our blog entries, this one is crammed full of detail. So, I would like to telegraph a few of the central take-home messages as I see them:
Our perception is that a big new area of longevity science may be opening up connected wih NAD, metabolisn and Sirtuins, one that may well offer new practical interventions that could lead to longer healthier lives. There are strong theoretical as well as experimental reasons for believing that enhancing body levels of NAD+ may have a chance of enhancing health and longevity while more conventional approaches do a limited job or fail. We identify those reasons here.
- There are several alternative approaches now being investigated to achieve such enhancement, including supplementation with NMN, or with nicotinamide riboside (NR) and direct IV infusion of NAD. We don’t know which one will be best. or even the extent to which any one really works in humans to enhance health and longevity. Nonetheless, we probably are seeing a commercial rush to bring NMN supplements to the market, and NR is already being sold as a supplement.
- There are additional theoretical reasons for suspecting that such benefits may not emerge or that they may require additional interventions for their realization. One approach for enhancing NAD+ levels may be far superior to the others. For example many of the benefits of NAD plus supplementation can be attributed to higher activity of the Surtuin SIRT1. However, what actually happens with Sirtuin expression is the result of a complex network of feedback-inhibition loops and many known factors can work to limit or eliminate Sirtuin levels or activity. We have very poor understanding of how these feedback inhibition loops ultimately net out in live mice under various real-life conditions, let alone in us.
- We offer many predictions below about probable health benefits of NAD+ supplementation. We need to acknowledge that some or all of these could be wrong.
- For this last reasons, it is very important to develop a panel of practically measurable bottom-line biomarkers that tell us what a form of NAD+ enhancement is actually doing in our body to enhance health and possible longevity. And, to tell us other things we don’t understand well, such as how best to synchronize NAD+ enhancement with our circadian rhythms and additional interventions such as fasting and exercise. Otherwise, we will be flying blind with respect to the efficacy of NAD+ enhancement, much as we have been traditionally flying blind with respect to the efficacy of consuming dietary health supplements. We identify a number of such possible biomarkers in the course of this blog entry, although we leave discussion of practical means to measure these biomarkers to a subsequent blog entry in this series.
The Discovery of NAD+ and NAD+ Metabolism
How NAD+ was discovered – Yeast fermentation of sugar
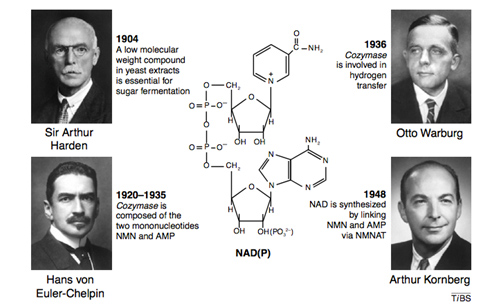
Four Nobel prize winners contributed to the discovery of NAD+, starting with Sir Arthur Harden who studied yeast fermentation of sugar back in 1904 and isolated a low molecular weight fraction and a high molecular weight fraction, both of which were necessary for the fermentation to occur. He did not know that the low molecular weight fraction was NAD+, but called this low molecular weight fraction “cozymaze”. Later on Hans von Euhler-Chelpin showed that cozymaze was made two mononucleotides, AMP and NMN. Then in 1936 Otto Warburg, showed that cozymaze was involved in hydrogen transfer (now called redox reactions). In 1948, Arthur Kornberg showed that NAD+ was synthesized enzymatically within cells by an enzyme that consumed ATP and linked NMN with AMP to form NAD+. All of these scientists won Nobel prizes during their career. However, it would be another 55 years before the primary structure of the NAD biosynthetic enzyme (NMNAT) was determined. Thus the entire discovery process of NAD+ and NAD+ biosynthesis took over 100 years to get to where we are today.
Popular but questionably effective downstream attempts to increase longevity and calorie restriction that actually does this
Next, again as background we review a) why antioxidants, vitamins, and hormones fail to reverse aging, b) the molecular pathways of calorie restriction (CR) which is the best known intervention for life extension, and c) how Sirtuins are the key molecular actors for health and longevity resulting from both calorie restriction and NAD+ enhancement.
Why Antioxidants, Vitamins, and Hormones failed to reverse aging – They treat the symptoms and not the causes
Although many drivers of human aging can be slowed or delayed, many of these factors are thought to be non-reversible reversible. (Ex: DNA gene mutations are not reversible). Most “anti-aging” supplements like anti-oxidants cannot reverse such aspects of aging. Instead, they merely “control damage,” and many have a mixed track record of efficacy in clinical trials. Likewise, hormone replacement therapy (HRT) does not reverse aging, despite the claims of some “anti-aging” clinics. In fact, emerging scientific studies have shown that exogenous anti-oxidant supplements and HRTs have a paradoxical effect. For instance, exercise has been shown to increase the expression of anti-oxidant genes and increase the expression of endogenous hormones (hGH, etc.). However, when exogenous anti-oxidants are used, this reduces the expression of anti-oxidant genes induced by the exercise producing a negative effect. Likewise, exogenous HRT use suppresses endogenous hormone production and thereby accelerates the decline in hormone gene expression that occurs with aging (via an epigenetic feedback-this is so good inhibition mechanism). This is why many testosterone users have testicular atrophy. Thus antioxidants and HRTs treat the symptoms (i.e. “downstream effects) of aging, rather than the cause (“upstream events”) of aging.
Current initiatives to restore NAD+ levels in individuals are attempts to affect what we believe are “upstream events” in aging to produce positive downstream events, such as already has been shown to be possible in various studies – like reversal of muscle aging in rodents.
Because nuclear NAD+ deficiency has so many impacts that mimic those of caloric restriction (CR), we look next at how it affects upstream events in molecular aging. We are also looking at CR for ideas for the best biomarkers for objectively evaluating the “upstream effects” of restoring NAD+ levels in the nucleus.
Why Calorie Restriction (CR) increases life span – CR affects the “upstream events” in molecular aging
Caloric restriction (CR) is the most scientifically-validated method of increasing life span and health span. This involves reducing caloric intake by 10-40% (below RDA) without inducing protein-calorie malnutrition. The molecular mechanisms of CR are complex, but simply put, they impede and slow “pro-aging pathways” and amplify “longevity pathways”. The two major “pro-aging pathways” are the Insulin/IGF-1 pathway and the mTOR pathway. CR inhibits both of these. The “longevity pathways” involve AMPK, Sirtuins, Glucagon, and Adiponectin. CR activates all of these. The diagram below shows the molecular mechanism of each of these factors in CR.

The Role of Sirtuins in Caloric Restriction (CR) – Sirtuins are integrally involved with all of the CR pathways and are the main actors for both calorie restriction and NAD+ health and longevity interventions.
In the diagram above, there are three red ovals that comprise a major molecular pathway of CR involving the family of enzymes called Sirtuins. One red ovals contain the symbols for the one of the 7 Sirtuin enzymes called Sirtuin 1 (SIRT1). Another red oval contains symbol for Nicotinamide adenine dinucleotide (NAD+), which is the substrate that activates all of the Sirtuin enzymes. Sirtuin enzymes consume NAD+, producing a by product called Nicotinamide (Nam), which inhibits SIRT enzymes. The last red oval represents the enzyme called Nampt, which is the rate-limiting enzyme in the pathway that converts Nam back into NAD+, thereby renewing the supply of NAD+ to activate SIRT enzymes. If you have read the Part 1 blog entry, you will recognize this pathway of regenerating NAD+ from Nam which is called the “NAD+ salvage pathway.” Activating these “red ovals” and restoring normal Sirtuin function is the goal of interventions intended to elevate or restore nulcear levels of NAD+.
The diagram above only shows one Sirtuin enzyme, SIRT1. In reality, there are 7 NAD+-dependent Sirtuins (SIRT1-7) which all play a role in CR molecular mechanisms. Sirtuins affect almost every oval in the diagram below because they activate or inhibit other CR proteins by removing acetyl groups from these CR proteins. In the diagram above, the small “Ac” stands for an “acetyl” group. SIRT1 directly affects all of the CR proteins that have an “Ac” on the diagram. It deacetylates and therefore tends to activate them.
For discussion of many of the benefits of Sirtuins, you can review the blogs on the series Slaying Two Dragons With One Stone (the Sound of Silence), Part 1, Part 2 and Part 3.
In addition to the major molecular pathway of CR on aging shown above, there are more important but lesser- known factors that change with CR, such as Klotho, p66shc, and DNA repair pathways. Although none of these pathways alone can account for the beneficial effects of CR, all of them probably work together to increase healthspan in all organisms studied to date. It is the involvement of NAD+ as a required substrate for the Sirtuin enzymes that is the focus of initiatives to restore the nuclear levels of NAD+.
Although the diagram does not illustrate this, NAD+ drives the Sirtuin-dependent deacetylation of many of the enzymes shown above, including eNOS, NF-kB, PGC-1a, Foxos, LKB1, and IRS1/IRS2. By NAD+-dependent deacetylation of these enzymes, Sirtuins thereby affect every major molecular pathway that involves aging. This is why many researchers believe restoring NAD+ levels in the cell is so important for affecting the “cause” of aging, rather than the “effects” of aging.
A 2012 review article, Cantó et. al. Targeting SIRT1 to improve metabolism: all you need is NAD+? anallyzes ” the pros and cons of the current strategies used to activate SIRT1 and explore the emerging evidence indicating that modulation of NAD+ levels could provide an effective way to achieve such goals.”
The Two Roles of NAD+ – Contrasting the Cofactor vs Signaling role of NAD+
(Note that the Part 1 blog entry introduces the main actors in the cast of NAD World, molecular entities mentioned below, like NAD+, NADH, NADPH. ATP. etc.)
NAD+ does two things: a) it serves as a cofactor in redox and important metabolic reactions in which case it is shuttled back and forth between the NAD+ and NADH forms but is not consumed, and b) it serves as a signaling molecule in which case it is consumed as a substrate.
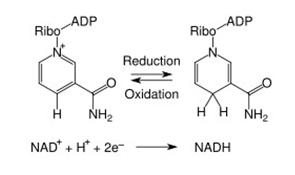
When NAD+ and NADP were discovered, they were thought to function only as a cofactor in redox reactions. Here they functioned as an “energy currency”, transferring high energy electrons from fuel oxidation (carbohydrates, fats, protein or alcohol) to the mitochondria (NADH) or the plasma membrane (NADPH) to generate ATP. In these reactions, NAD(P) “accepted” hydride groups (2 electrons and one proton) and NAD(P)H “donated” hydride groups, transferring the high energy electrons to another substrate. Thus NAD/NADH was the recycled cofactor pair in the catabolic oxidation of carbohydrates, fatty acids, proteins, and alcohol. On the other hand, NADP/NADPH was the recycled cofactor pair in the anabolic synthesis of fatty acids and cholesterol. In these redox reactions, no NAD or NADP molecules are consumed. Over 200 cellular enzymes utilize either NAD+/NADH or NADP/NADPH as a cofactor this way.
It was not until recently, however, that a second role for NAD+ as a signaling molecule was discovered. In these reactions, NAD+ does not accept hydride groups and the molecule is not recycled. Instead, the NAD+ molecule is consumed as a substrate and the glycosidic bond between the ADP-ribose moiety and the nicotinamide moiety (NAM) of the molecule is broken, releasing NADM and ADP-ribose as byproducts of the reaction. Sirtuins are just one of many enzymes that “consume” NAD+ as a substrate. The two contrasting roles for NAD+ as a cofactor vs a substrate are illustrated below:
The Redox/Co-Factor Role of NAD+ and NADPH and The Signaling Compound/Substrate Role of NAD+
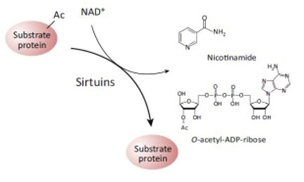
What makes and what eats-up NAD+? Causes of NAD+ deficiency with aging.
Although NAD+ can be made from the amino acid tryptophan by an 8-step pathway called the de novo pathway, this pathway does not work very well in most cells. For this reason, most of the consumed NAD+ is converted to nicotinamide and then “salvaged” by a two step process that converts it back into NAD+. This pathway is called the salvage pathway and is rate-limited by the first enzyme in the two-step pathway called NAMPT. NAMPT gene expression can be activated by caloric restriction (CR) or exercise and this may be a major reason why both CR and exercise have long term health benefits. However even with exercise and caloric restriction, nuclear NAD+ levels decline with aging.
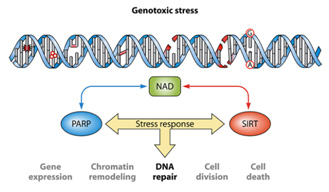
It is unclear now if the cause of age-related nuclear NAD+ deficiency is due to Sirtuins, PARPs, CD38, or a decline in the expression of NAMPT. Most studies suggest that it is a combination of all four factors and that “genotoxic stress” is a major driving force for nuclear NAD+ deficiency. The synergistic effects of genotoxic stress inducing single stranded DNA breaks and double-stranded DNA breaks and the combined effect of Sirtuins and PARPs is illustrated below:
Also, NAD+ can be provided by an exogenous source such as an IV, and its level can possibly enhanced such as by NMN or NR supplementation as discussed below. And also as outlined below numerous factors can affect SIRT activation levels.
NAD+ and aging
Nuclear NAD+ decline and Aging – One cause of aging is thought to be the effect of NAD+-consuming enzymes in the cell nucleus
One of the “upstream events” in aging appears to be the decline in NAD+ within the nucleus of the cell. This appears to occur within all 37 trillion cells within the human body. The decline is due to several classes of NAD+-consuming enzymes called Sirtuins, PARPs, and ADP-ribose glycohydrolases. These enzymes perform absolutely essential functions required for cell health and each consumes NAD+ as a substrate. Aging appears to be associated with increasing insufficiency of enough NAD+ to support the growing demands of these. Sirtuins are a class of NAD+-consuming enzymes needed for double stranded DNA repair, for epigenetic gene silencing, and for chromatin remodeling. Poly-ADP-ribose Polymerases (PARPs) is another class of NAD+-consuming enzymes involved with repair of single stranded break DNA damage. PARPs also initiate cell death (apoptosis) in cancer cells and cells whose DNA is beyond repair. More recently, a third type of NAD+-consuming enzyme has been found in the nucleus called CD38, once thought to only exist outside the cell. CD38 is a part of the family of enzymes called ADP-ribose glycohydrolases. Thus there are at least three families of enzymes all competing for the same pool of NAD+ within the nucleus of the cell.
Aging and NAD+ – Aging in the NAD world-view is due to a “nuclear NAD+ deficiency” incurred from NAD+-consuming enzymes
For quite some time, caloric restriction (CR) has been regarded as the most scientifically-validated method to retard aging. CR research has lead to the discovery of several major molecular pathways that accelerate or retard aging, including the Insulin/IGF-1 pathway (age accelerator), the mTOR pathway (age accelerator), the AMPK pathway (aging brake), and Sirtuins (aging brake). It is the role of NAD+ as a required substrate for Sirtuins that lead to the recent discovery that there is a nuclear NAD+ deficiency that affects not only Sirtuins, but another set of important NAD+ consuming enzymes called Poly-ADP-ribose Polymerases (PARPs). Both the Sirtuins and the PARPs are involved with the DNA damage response to genotoxic stress. Sirtuins play a major role in double-stranded DNA breaks whereas PARPs play a major role in single-stranded DNA breaks. Here is a simplified diagram showing how both SIRTs and PARPs use up NAD+ in the nucleus of the cell:
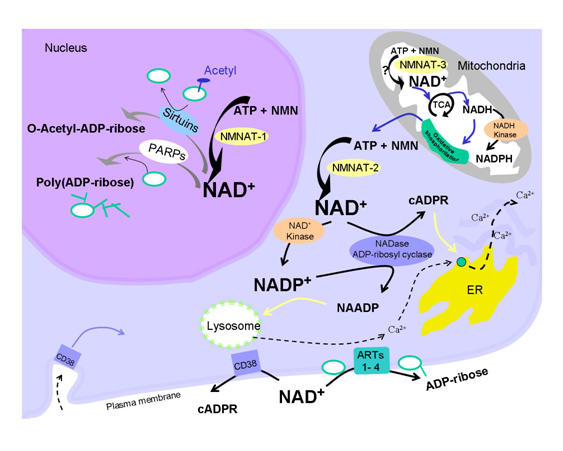
Restoring nuclear NAD+ levels may reverse aging
Recent work has shown that restoring nuclear NAD+ levels in aging mice actually reversed a common phenotypes of aging – that of mitochondrial dysfunction. Mitochondrial dysfunction manifests itself as a decline in ATP production, a decline in heat production, excessive cellular free radical production, and increased DNA damage.
Although there are many proposed strategies for restoring nuclear NAD+ levels to normal, the only one that has been successfully accomplished this experimentally so far (with mice) is nicotinamide mononucleotide (NMN). However, many experts believe that supplementation with nicotinamide riboside (NR) or directly with NAD will also restore nuclear NAD+ levels. See the 2012 publication Canto et. al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet induced obesity. “As NAD+ is a rate-limiting cosubstrate for the sirtuin enzymes, its modulation is emerging as a valuable tool to regulate sirtuin function and, consequently, oxidative metabolism. In line with this premise, decreased activity of PARP-1 or CD38—both NAD+ consumers—increases NAD+ bioavailability, resulting in SIRT1 activation and protection against metabolic disease. Here we evaluated whether similar effects could be achieved by increasing the supply of nicotinamide riboside (NR), a recently described natural NAD+ precursor with the ability to increase NAD+ levels, Sir2-dependent gene silencing, and replicative life span in yeast. We show that NR supplementation in mammalian cells and mouse tissues increases NAD+ levels and activates SIRT1 and SIRT3, culminating in enhanced oxidative metabolism and protection against high-fat diet-induced metabolic abnormalities. Consequently, our results indicate that the natural vitamin NR could be used as a nutritional supplement to ameliorate metabolic and age-related disorders characterized by defective mitochondrial function.”
Others have suggested alternative strategies such as PARP inhibition, CD38 inhibition, or increasing NAMPT activity. We are interested in all of the above. We are not interested in limiting our approach to one particular strategy. Likewise, we do not believe that any particular NAD precursor, NAD metabolite, or enzyme inhibitor is a replacement for sleep, exercise, and caloric restriction, which have already been scientifically proven to help ameliorate the nuclear NAD+ problem. Rather, we are interested in doing whatever it takes to restore nuclear NAD+ levels, using sleep, exercise, and CR as fundamental strategies and various NAD precursors, PARP inhibitors, and CD38 inhibitors as adjuncts to a healthy lifestyle. Further, we strongly suspect that determination of what works best and under what conditions cannot be based on theory and will require monitoring a panel of biomarkers. My personal goal is not to increase lifespan but to increase healthspan, although increasing healthspan could increase lifespan.
This diagram projects an increase of lifespan possibly associated with NMN
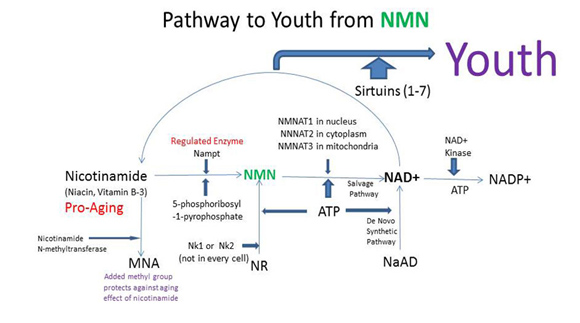
How NAD+ activates SIRTs and the competing “NAD+ consumers”
Based on the evidence from CR studies, there is strong scientific evidence that activating Sirtuins has major positive health effects and longevity benefits in most all animal models. Although there are 7 different Sirtuin enzymes (SIRT1-7), SIRT1 is the most well understood member of this family of enzymes. All 7 Sirtuins require NAD+ as a substrate for the enzymes to work. Sirtuin enzymes consume NAD+, converting it into nicotinamide (NAM). NAM itself inhibits SIRT1 activity, but NAM can be “salvaged” and converted back into NAD+ (see diagram below). This conversion of NAM back into NAD+ is a two-step enzymatic process called the “NAD+ salvage pathway” described in our Part 1 blog entry and covered more in additional detail here.
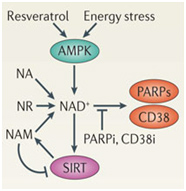
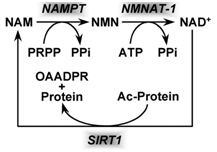
Image source NAD+ is consumed by enzymatic activity of all 7 of the Sirtuin isoforms (SIRT1-7). The consumption of NAD+ produces the byproduct, Nicotinamide (NAM) which can be “recycled” to NAD+ in the 2-step process shown above that is called the salvage cycle, which requires PRPP & ATP to run the cycle.
As discussed in the Part 1 blog entry, there are other enzymes that also require NAD+ as a substrate, such as the PARP family of enzymes as well as CD38 (see diagram below). These other NAD+-consuming enzymes “compete” with SIRT enzymes in the nucleus and this competition may be the primary reason why nuclear NAD+ levels decline with aging. Again, this is why restoring nuclear NAD+ levels is thought to be a good idea.
The above two diagrams illustrate the key “suspects” in the etiology of SIRT deficiency-induced aging. As you can see, this “SIRT1 deficiency” is not a deficiency in the protein, but a deficiency in the activity of the protein. It has been clearly shown that in most scenarios, there is no decline in SIRT1 protein availability with aging. This may be because the SIRT1 protein can regulate its own gene expression. This auto-regulation may be why the expression of SIRT1 protein does not decline with aging.
What is also a mystery is that aging also is associated with a decline in the expression of SIRT3. It appears that the reason for this decline in SIRT3 expression may be due to the fact that SIRT1 regulates the gene expression of SIRT3. This is why many experts believe that the decline in SIRT1 activity is an “upstream event”, whereas the decline in SIRT3 is a “down stream events”. The following possible causes for the decline in SIRT1 activity are illustrated in the diagrams above and listed in the table below:
Possible Causes of the Decline in SIRT1 activity with Aging1.
Decline in iNAMPT activity – The NAD+ salvage pathway has a rate-limiting enzyme called Nicotinamide phosphoribosyltransferase (NAMPT). Because there is both an intracellular and an extracellular localization of NAMPT, it is sometimes abbreviated as iNAMPT (intracellular NAMPT) and eNAMPT (extracellular form, which has also been called eNAMPT, PBEF, or Visfatin). eNAMP circulates in the blood and will be discussed elsewhere. With iNAMPT, several independent studies have shown that iNAMPT gene expression and iNAMPT protein activity declines with aging and that this decline in iNAMPT activity precedes replicative senescence of cells, such as human vascular smooth muscle cells (VSMCs) (Eric van der Veer, Extension of Human Cell Lifespan by Nicotinamide Phosphoribosyltransferase 2007). Even more interestingly, before the VSMCs became senescent, the decrease in NAMPT enzymatic activity was even greater than the decline in NAMPT gene expression with NAMPT enzyme activity dropping to 14% of baseline prior to VSMC senescence. This is a very powerful argument that NAMPT is a “longevity protein” that extends the lifespan of VSMCs by activating SIRT1 and restraining the accumulation of p53 (SIRT1 mediates p53 degradation).
The “good news” is that there are 4 ways to increase NAMPT activity: caloric restriction (CR), fasting, exercise, and ATR1 blockade. The mechanism by which CR and fasting increase NAMPT is probably due to the fact that the SIRT1 protein regulates the NAMPT gene (Zhang, Enzymes in the NAD+ Salvage Pathway Regulate SIRT1 Activity at Target Gene Promoters 2009). The mechanism by which exercise induces an increase in NAMPT expression is via AMPK-mediated PGC-1α activity. Athletes have a 2-fold higher levels of NAMPT in their skeletal muscle compared with sedentary adults (obese, non-obese, and T2DM). Three weeks of exercise training increases NAMPT protein in skeletal muscles by 127%.
AICAR, a potent “exercise mimetic”, increases skeletal muscle NAMPT mRNA by 3.4-fold. (Costford et.al., Skeletal muscle NAMPT is induced by exercise in humans, 2010). Recently, a fourth method to increase iNAMPT gene expression was found: angiotensin receptor 1 (ATR1) blockade. ATR1 blockers such as candesartan and telmisartan increase expression of NAMPT and SIRT3 genes, increased NAD+ levels, increased mitochondrial biogenesis, and reduced atherosclersosis in in vivo studies. Even more surprising was that in ATR1 gene knock-out mice, there was a 26% longer lifespan. (Benigni et.al., Disruption of the Ang II type 1 receptor promotes longevity in mice 2009). This effect was not mediated by the usual CR pathways, but appeared to be a CR independent mechanism, mediated by an increase in iNAMPT and SIRT3.
Conclusion: CR, fasting, and regular exercise decelerates the deleterious effects of aging via SIRT1-dependent pathways by stimulating of NAD+ biosynthesis via increase in NAMPT gene expression. ATR1 blockade increases NAMPT gene expression via an unknown, CR-independent mechanism.
- Increase in PARP activity – There is recent evidence that a decline in NAD+ levels occurs in the nucleus of the cell with aging. As we have already indicted, the exact cause of this “nuclear NAD+ decline” is unknown and is the focus of research initiatives for restoration of nuclear NAD+. We have already discusssed the role of the PARPs as NAD+ consumers above and in the Part 1 blog entry.
SIRT1 – The Most Important Sirtuin in Aging and Age-related Diseases
Here, we review some of the most compelling reasons why SIRT1, the queen of the Sirtuins, is so important for health and longevity.
Depletion of SIRT1 by siRNA knockdown significantly alters the expression of about 200 genes (Zhang, Enzymes in the NAD+ Salvage Pathway Regulate SIRT1 Activity at Target Gene Promoters 2009). Here are some of the effects of SIRT1 on various organs in the body that have been scientifically proven in animal models. We believe that restoring nuclear NAD+ levels to normal will probably have all of these effects on the body, as well as many other positive effects, since NAD+ activates all 7 of the Sirtuins, not just the SIRT1 isoform.
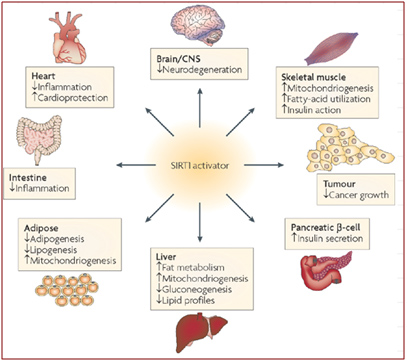
Image source: Sirtuins — novel therapeutic targets to treat age-associated diseases
As you can see from the diagram above, there are beneficial effects of SIRT1 activation on almost every organ system in the body. Many of these have to do with ameliorating the signs or symptoms of metabolic syndrome and type II diabetes mellitus. For those that are not familiar with Sirtuin proteins, it is important to understand that SIRT1 does not circulate as a hormone, but instead, is a protein largely confined to the nucleus of the individual cell. In the cell nucleus, SIRT1 deacetylates many proteins that activate or inhibit gene expression. These effects are thought to be largely but not exclusively due to epigenetic mechanisms involving the deacetylation of histone proteins. SIRT1 deacetylates many other proteins found in the nucleus other than histone proteins. The following sections will cover both the histone and the “non-histone” effects of SRIT1 in the nucleus of the cell. All of these benefits of SIRT1 are easier to understand if one familiarizes themselves with the fundamental aspects of epigenetics. This article does not go into detail about epigenetics, but it would be wise for the unfamiliar reader to delve into this topic first before proceeding to read the following sections. Past blog entries related to epigenetics are listed here. And the many prevous blog entries related to Sirtuins are listed here. For now, we will focus on the specific effects of SIRT1 within the nucleus of a cell, as described below. Then we’ll cover the other nuclear-localized members of the Sirtuin family, SIRT6 and SIRT7.
Major SIRT1 Functions #1: Preventing cellular senescence –SIRT1 prevents cell cycle arrest via six mechanisms. We first list these mechanisms and then review what cellular senescence is and further discuss why it is important, and what these SIRT1 impacts are.
1) SIRT1 helps repair double stranded DNA breaks, which is a major “upstream cause” of cellular senescence
2) SIRT1 epigenetically prevents expression of the p16INK4a/ARF promoter by maintaining promoter hypermethylation
3) SIRT1 epigenetically prevents gene expression of p16INK4a/ARF decetylation of H3K9 histone proteins
4) SIRT1 genetically activates Akt/p70S6K1 signaling, thereby inducing p16INK4a/ARF repression
5) SIRT1 prevents p53-induced cell cycle arrest by deacetylating p53
6) SIRT1 prevents the formation of heterochromatin by deacetylating histone linker proteins (H1K24)
About cellular senescence
Cellular senescence is defined as “cell cycle arrest” and is a major hallmark of aging in all species. In old age, approximately 15-20% of cells in non-human primates show markers for cellular senescence (Herbig, Cellular Senescence in Aging Primates 2006). In human bone marrow stromal cells cultured in vitro, the percentage of senescent cells increases 4% per population doubling in old bone marrow cells vs only 0.4% per population doubling in young bone marrow cells (Stenderup, Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells 2003).
Some past blog entries discussing cell senescence and the roles of p16INK4a/ARF are listed here.
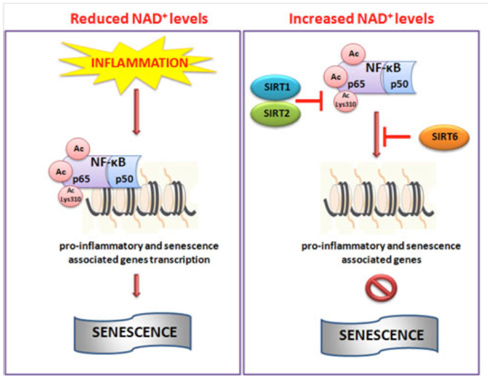
Image and legend source: Cellular Repair and Reversal of Aging: the Role of NAD “Schematic illustration of NAD+-mediated sirtuins actions on NF-κB. During inflammation reduced levels of NAD+ do not impaired the hyperacetylation of NF-κB which in turn through promoter region of target genes activate pro-inflammatory pathway and senescence. Conversely, increased cellular levels of NAD+ activate SIRT1, 2 and 6 which deacetylate NF-κB inhibiting its transcriptional role and then inflammation.”
Although cell senescence has many beneficial, vital roles in normal life (embryogenesis, wound healing, cancer prevention, red blood cell aging, etc.) senescent cells are rare in young organisms. In old age, all species start to accumulate old cells that will not divide (i.e. are senescent). Normally old cells undergo apoptosis, but these senescent cells are resistant to apoptosis, which means they hang around for longer than cells should. Moreover, they secrete a toxic mixture of secretions that are pro-inflammatory and actually help cancers grow (called the senescence associated secretory phenotype, aka SASP). Other than genetic modifications of cells, no “cure” for the accumulation of senescent cells has been found to date. For this reason, many researchers have believed the best way to prevent aging would be to prevent cellular senescence without increasing the risk of cancer. Caloric restriction (CR) is the most effective method to date that has been discovered for preventing cellular senescence. SIRT1 appears to be an important reason why CR prevents cellular senescence.
To understand cellular senescence on a molecular level, it is important to understand the major causes of cellular senescence, which includes DNA damage and telomere shortening. When either of these events occur, three key proteins trigger cell cycle arrest. They are p16INK4a protein, the ARF protein, and the p53 protein. Since p16INK4a and ARF are both transcribed from the same gene, I will discuss them together here. However, I will briefly discuss DNA damage and telomere shortening first.
The initial discovery of the phenomena of cellular senescence was by Leonard Hayflick when he noted that diploid fibroblasts in culture would only keep dividing for a finite number of “population doublings”, and then would stop dividing. The number of cell divisions that would trigger this phenomena was referred to as the “Hayflick limit”, but the cause was unknown until researchers discovered that telomeres shortened with each cell division due to the DNA “end replication problem”. Later on, an enzyme called telomerease was discovered that could counteract the telomere shortening that occurred with cell divisions. The researchers who discovered telomerase won the Nobel prize in 2009, but since then, many other factors have been discovered that regulate telomere length (Ex: oxidative stress, the lncRNA called TERRA, Shelterin protein caps on the telomeres that protect the ends, Rap1, Tankyrases, SIRT6, histone protein silencing of telomeric DNA, etc.).
Ten years ago, many of us concerned with aging thought that since telomeres usually get shorter with age and shorter telomeres can lead to cell senescence, health and longevity could be enhanced by interventions that make telomeres longer. That was a very exciting idea, but it turned out to be wrong. We now know that telomere lengths are mostly downstream responses to upstream events, just like grey hair and wrinkles are downstream consequences of aging. See the blog entries Nuclear Aging: The View from the Telomere end of the Chromosome – Part 1 – context, history, and about telomere lengths and Part 2 – Telomere Molecular Biology and many other blog entries we have writtem about telomeres and telomerase for details. There is just a possibility that we could be equally wrong about our new exciting idea – that of enhancing NAD+ as a health and longevity intervention. Time will tell.
When telomeres shorten to a critical point, they trigger the DNA damage response (DDR), which activates p53 and can induce cellular senescence. Later on, it was discovered that even non-dividing cells and cells with longer telomeres could undergo cellular senescence. For instance, X-ray or gamma ray radiation can induce cellular senescence in cells with longer telomeres. Likewise, oxidative stress from intrinsically-produced ROS or exposure to oxidative stress by external ROS (Ex: exogenous H2O2) can also trigger cellular senescence. In these cases, the cause for the cellular senescence appears to be DNA damage. Double stranded DNA breaks (DSBs) appear to be the most closely associated with cellular senescence, regardless of whether the DSBs were induced by ionizing radiation (X-rays, UV light, etc.), DNA damaging drugs (chemotherapy), or oxidative stress (endogenously generated or exogenously applied).
A key factor in cellular senescence is the increased expression of two proteins that trigger cellular senescence, p16INK4a and ARF. These two proteins are actually part of the same gene (i.e. they share exons) but because they are encoded in different reading frames, they have different amino acid sequences and different functions. As a result of this “shared gene”, they are often referred to as the “INK4a/ARF locus”. p16INK4a is an inhibitor of the cyclin-dependent kinases CDK4 and CDK6. ARF regulates p53 stability through inactivation of the p53-degrading ubiquitin ligase, MDM2. In young cells and young organisms, the INK4a/ARF locus is epigenetically repressed by Polycomb proteins, but in old age, the INK4a/ARF locus is “derepressed”, the gene is transcribed, and production of these two proteins increases by as much as 42-fold in different organs in older mice (Krishnamurthy, Ink4a/Arf expression is a biomarker of aging 2004). In this context, the activities of SIRT1 listed above (Numbers 2, 3, and 4) mitigate against cell senescence by repressing p16INK4a/ARF or its effects.
Interestingly, calorie restriction retards the increase in INK4a/ARF protein accumulation in all organs, reducing the age-associated increase in INK4a/ARF proteins by 2 and 16-fold. (Also Krishnamurthy, Ink4a/Arf expression is a biomarker of aging 2004). Although the loss of Polycomb protein repression is a major reason for the increase in INK4a/ARF proteins, decline of SIRT1 expression plays a major role here as well. For instance, glucose restriction (GR) has been found to increase SIRT1 mRNA levels and SIRT1 protein levels within cells and to inhibit p16INK4a/ARF expression. As a result, cellular lifespan was found to be dramatically increased (by 4 weeks) in normal cells with GR, whereas the same glucose restriction increased apoptosis (18%) in immortalized cells causing them to die (Li, Tollefsbol, Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control ofhTERT and p16 expression 2010)(Li, Tollefsbol, p16INK4a Suppression by Glucose Restriction Contributes to Human Cellular Lifespan Extension through SIRT1-Mediated Epigenetic and Genetic Mechanisms 2011). When the molecular mechanisms were studied on the role of GR in repressing p16INK4a/ARF expression, several mechanisms were found to play a role as follows:
1. Promoter site DNA (CpG) methylation
Glucose restriction induced hypermethylation of the p16INK4a/ARF promoter, which then prevents binding of the E2F-1 transcription factor. This results in down-regulation of the p16INK4a/ARF locus, preventing cell apoptosis and cellular senescence, which leads to increased cell lifespan. (Li, et.al. as above, 2010). Recently, other researchers have shown that SIRT1 can deacetylate DNA methyltransferase 1(DNMT1) and that compared to other HDACs, SIRT1 is the most robust deacetylator of DNMT1 (Peng, SIRT1 Deacetylates the DNA Methyltransferase 1 (DNMT1) Protein and Alters Its Activities 2011). “In summary, we show that DNMT1 is acetylated at multiple lysines and that SIRT1 deacetylates DNMT1 in vitro and in vivo. Deacetylation of DNMT1 at specific lysines enhanced its methyltransferase activity, changed its transcription repression activity and cell cycle regulatory function, and impaired its capacity to silence TSGs. In contrast to class I HDACs, which boost the silencing effect of DNMT1 by chromatin modification or stabilization of DNMT1 (59, 81), SIRT1 directly modifies DNMT1 activity.”
The same authors showed that nicotinamide, an inhibitor of SIRT1, reduced DNA promotor methylation by DNMT1. Since the main role of DNMT1 is to maintain CpG methylation (as opposed to de novo methylation), Thus it is likely that SIRT1 plays a role in maintaining DNA promoter site (CpG) methylation of the p16 promoter, although this mechanism not yet been specifically proven for the INK4a/ARF locus. Again, the key point of interest here is that supporting methylation and therefore inactivation of ther p16 promoter site is a likely action of SIRT1 for countering age-related cell senescence.
- Promoter site Histone deacetylation
The best way to understand the role of SIRT1 in cellular senescence is to understand the molecular mechanisms of how Sirtuin enzymes work. Although Sirtuins do have other molecular functions, their primary job is to remove acetyl groups from lysine amino acids on proteins. In the nucleus, SIRT1 removes acetyl groups (i.e. deacetylation) from many proteins, but the major target of SIRT1 are histone proteins (H1, H3, and H4). Histone proteins make up the “spools” (called nucleosomes) which DNA is wound around. When the spools are tightly compacted together, genes cannot be transcribed from the DNA wound around the nucleosomes. This chromatin compacted state is called “heterochromatin”. When the nucleosomes are spread out and not compacted, genes can be transcribed from the DNA and this chromatin state is called “euchromatin”. SIRT1 plays a major role in determining if chromatin is in the heterochromatin vs the euchromatin state. Specifically, SIRT1 rmoves an acetyl group from histone subunit 1 (abbreviated as H1K24). When this occurs, the chromatin remains in the euchromatin state and can be transcribed. When there is a nuclear deficiency of NAD+, H1K24 becomes acetylated and chromatin compaction (heterochromatin) occurs. The formation of heterochromatin is a hallmark of cellular senescence and aging. This is illustrated in the diagram below. On a molecular level, there is no clearer “upstream explanation for aging” than this diagramt, illustrating what happens in young cells when NAD+ levels are high (SIRT1 activation) and what happens in old age when nicotinamide levels are high (SIRT1 inhibition). The 4 main SIRT1 anti-aging effects involve protein deacetylation in the nucleus:
1) deacetylation of the histone linker protein, H1;
2) deacetylation of the tumor suppressor, p53;
3) deacetylation of histone H4 at lysine 16 and
4) deacetylation of histone H3 at lysine 9.
When H1 is deacetylated, chromatin is relaxed and gene expression occurs (euchromatin). If p53 is deacetylated, it cell survival occurs and cell cycle arrest does not occur (i.e. senescence). When H4K16 is deacetylated, genes can be silenced. With low levels of nuclear NAD+,
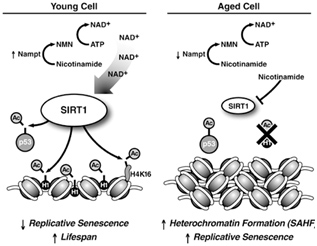
Image and legend source: Sirtuins: critical regulators at the crossroads between cancer and aging “SIRT1 expression and activity decrease during aging. High levels of SIRT1 protein in young cells in combination with high levels of nicotinamide adenine dinucleotide lead to deacetylation of p53 and histone proteins to promote longevity (left panel). SIRT1 levels are lower in aged cells, and higher levels of nicotinamide inhibit SIRT1 activity (right panel). The resulting hyperacetylation of p53 can induce replicative senescence. Deacetylation of histone H1 leads to its degradation that promotes formation of senescence-associated heterochromatic foci (SAHFs). Failure to downregulate SIRT1 during aging may promote cell survival after oxidative damage, leading to the accumulation of mutations, and an increased risk of cancer development.”
When there is inadequate SIRT1, H1K24 gets acetylated and the chromatin becomes compacted, forming “senescence associated heterochromatin” (SAH). SAH is a major cellular marker of aging. Thus the loss of SIRT1 H1K24 deacetylation directly causes this aspect of aging. However, if SIRT1 H2K24 deacetylation activity is maintained in old age, then SIRT1 becomes a “longevity protein”. Unfortunately, with aging, there is a nuclear deficiency of NAD+. As a result, SIRT1 cannot deacetylate H1K24 and senescent cells accumulate.
Note a point of clarification here on the different contexts for and meanings of deacetylation. The specific histone H1 relates to links between spindles of DNA, Keeping it deacetylated via SIRT1 results in uncompacted chromatin which facilitates ready gene expression. When it comes to histones H3 and H4, acetylation has the opposite effect; it locally uncompacts the chromatin and allows ready gene expression. And deacetylation inhibits gene expression in these cases, So, H3 and H4 deacetylation is very useful for silencing pro-aging genes such as senescence-associated ones.
Major SIRT1 Functions #2: Gene silencing – H3K9 and H4K16 deacetylation in the nucleus
Two histone proteins that are deacetylated by SIRT1 are histone subunit 3 (H3) and subunit 4 (H4) . SIRT1 removes an acetyl group from a specific lysine (K9) on H3 and a specific lysine (K16) on H4. H3K9 is a “site-specific” deacetylase target of SIRT1 and SIRT6, whereas H4K16 is a “site-specific” deacetylase target of only SIRT1. Both H3K9 and H4K16 deacetylation of histones have the effect of silencing genes as mentioned above. See the discussion and the diagrams in the blog entry Slaying Two Dragons with the Sound of Silence: – How to Keep Your Repetitive DNA Turned Off with “3 Songs”: Sirtuins, Polycomb Proteins, and DNMT3.
The major (non-histone) “longevity mechanism” is the effect of SIRT1 on p53, also shown below.
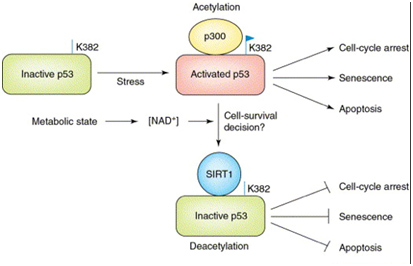
In the diagram above, SIRT1 deacetyates the p53 protein thereby reducing its transcriptional activity. p53 is the master “guardian of the genome”, and serves as a tumor suppressor to prevent cancer formation by shutting off the cell cycle in damaged cells. So, turning it off in cancer cells is not a good idea. p53 also increases cell survival in non-cancerous cells and also activates apoptosis when the cells are old or too damaged to repair.
From the 2012 publication Current advances in the synthesis and antitumoral activity of SIRT1-2 inhibitors by modulation of p53 and pro-apoptotic proteins” — As sirtuins are involved in many physiological and pathological processes, their activity has been associated with different human diseases, including cancer. Especially two sirtuin members, SIRT1 and SIRT2, have been found to antagonize p53-dependent transcriptional activation and apoptosis in response to DNA damage by catalyzing p53 deacetylation. The findings that SIRT1 levels are increased in a number of tumors highlight the oncogenic role of sirtuins, in particular, in the down-modulation of p53 oncosuppressor activity. Along this lane, cancers carrying wild-type (wt) p53 protein are known to deregulate its activity by other mechanisms. Therefore, inhibition of SIRT1 and SIRT2, aimed at restoring wt-p53 transcriptional activity in tumors that retain the ability to express normal p53, might represent a valid therapeutic cancer approach specially when combined with standard therapies.”
In addition, SIRT1 deacetylates a specific site on the histone 4 subunit of the nucleosomes, which are the spools on which DNA is wound. When SIRT1 deacetylates lysine at position 16 on the histone 4 protein (H4K16), this silences the gene on the DNA wound around this histone. Thus SIRT1 has multiple effects all due to its ability to remove acetyl groups from proteins. These are all dependent on the nuclear levels of NAD+ within the cell.
Image and legend source: Sirtuins: critical regulators at the crossroads between cancer and aging “ SIRT1 expression and activity decrease during aging. High levels of SIRT1 protein in young cells in combination with high levels of nicotinamide adenine dinucleotide lead to deacetylation of p53 and histone proteins to promote longevity (left panel). SIRT1 levels are lower in aged cells, and higher levels of nicotinamide inhibit SIRT1 activity (right panel). The resulting hyperacetylation of p53 can induce replicative senescence. Deacetylation of histone H1 leads to its degradation that promotes formation of senescence-associated heterochromatic foci (SAHFs). Failure to downregulate SIRT1 during aging may promote cell survival after oxidative damage, leading to the accumulation of mutations, and an increased risk of cancer development.”
Major SIRT1 Functions – #3: Circadian Clock Control – keeping the clocks in every cell regulated
We discussed the role of SIRT1 in controlling circadian clocks in the Part 1 blog entry, Also, you can see the blog entries Circadian Regulation,NMN, Preventing Diabetes, and Longevity and Shedding new light on circadian rhythms. Pathways involved are illustrated in this diagram:
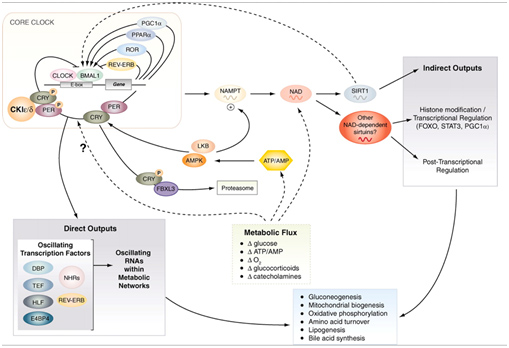
Image source: Circadian integration of metabolism and energetics
Major SIRT1 Functions – #4 : Reducing Inflammation – inhibition of both glucose and fatty acid-induced inflammation.
Inflammation and aging seem to go hand-in-hand, a process that has been referred to as “inflammaging”. Caloric restriction can reverse a significant amount of this inflammaging as a result of both the reduction in insulin/IGF-1 pathway signaling and also via a reduction in free fatty acid-induced inflammation. Sirtuin activation associated with CR has been shown to have a major role in CR as a molecular mediator of this effect on inflammaging. Here are some of the ways this occurs:
SIRT1 and Glucose-mediated inflammaging – Insulin resistance is the hallmark of this pathway of inflammaging.
SIRT1 improves insulin sensitivity in insulin resistant states by inhibiting protein tyrosine phosphatase 1B (PTP1B), which is a direct intracellular inhibitor of insulin action (This effect is only seen in insulin resistant states, however, and not in insulin sensitive states). In addition, SIRT1 increases the circulating plasma hormone that is made in fat, called adiponectin, which increases insulin sensitivity. Adiponectin also directly inhibits TNF-α and the conversion of macrophages to foam cells. This reduces adhesion molecules and the number of macrophages attached to endothelial cells. These SIRT1-induced effects reduce the atherosclerotic effects of insulin, high sugar diet, and high fat diets.
Probably the greatest positive effect of SIRT1 on inflammaging is by its inhibition of NF-kβ signaling. Normally, insulin signaling increases inflammation via Akt-mediated activation of the “master inflammatory switch”, NF-kβ, a transcription factor that turns on all of the major inflammatory genes. However, when SIRT1 activity is increased, SIRT1 deacetylates NF-kβ and this inactivates the transcription factor. As a result, all of the NF-kβ controlled inflammatory genes show less expression, including TNF-α, IL-6, C-reactive protein, and the cJun N-terminal Kinase (JNK) transcription factor. The diagram below illustrates this well:
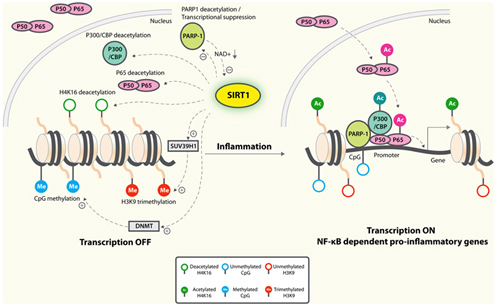
Image and legend source: Sirtuins in neurodegenerative diseases: an update on potential mechanisms ” Anti-inflammatory mechanisms of SIRT1. SIRT1 deacetylates p65 and blocks the transactivation of NF-κB-dependent gene expression. SIRT1 suppresses the activity of PARP-1, a coactivator of NF-κB-dependent transcription, by deacetylation and by inhibiting its expression. PARP-1 activation could deplete NAD+, resulting in inhibition of SIRT1 and NF-κB activation. On the epigenetic level, SIRT1 represses NF-κB-dependent inflammatory gene expression by deacetylating H4K16 and also by recruiting more components of repressor complexes. SIRT1 deacetylates and activates histone methyltransferase SUV39H1, which suppresses expression of inducible inflammatory genes. DNA methylation is associated with suppressed expression.”
In summary, SIRT1 has a net anti-inflammatory effect in glucose-mediated inflammaging and increases insulin sensitivity in insulin-resistant conditions, but not in baseline conditions of insulin-sensitive states.
SIRT1 and Fatty acid-mediated inflammaging – High fat diets have also been shown to induce inflammaging. Here the pro-inflammatory effects are independent from insulin and are mediated by intracellular free fatty acid overload within cells. These high free fatty acid levels inhibit the forkhead transcription factor, FOXO1. As a result of FOXO1 inhibition, there is an increase in ROS, IL-6, PAI-1, and MCP-1 gene expression. SIRT1 increases adiponectin levels which in turn, activates FOXO1 activity and increases the interaction between FOXO1 and C/EBPβ transcription factors. In summary, SIRT1 has a net anti-inflammatory effect in fatty-acid mediated inflammaging and this effect is mediated primarily by adiponectin and the FOXO1 transcription factor.
Conclusion: Restoring NAD+ levels to normal in the nucleus of cells should increase insulin sensitivity, increase adiponectin levels, and reduce inflammation induced by both a high glucose and high fat diet. We predict that biomarkers for insulin sensitivity (fasting blood sugar, 2-hour glucose tolerance testing, fasting insulin, 2-hour insulin tolerance testing, HOMA2, HOMA-IR, and Glycomark tests will improve with increases of nuclear NAD+, but only in insulin resistant states. We also predict that biomarkers for inflammation will decline with such nuclear NAD+ increases, especially adiponectin (increase), TNF-α (decrease), IL-1β (decrease), IL-6 (decrease), IL-8 (decrease), andC-reactive protein (decrease). However, if these values are normal in baseline conditions in a person undertaking to increase their nuclear NAD+, no change in these numbers will be evident.
Major SIRT1 functions: #5 – DNA repair – It is a key enzyme in repairing double stranded DNA breaks (DSBs)
Homologous recombination is the highest-fidelity mechanism for repairing DSCs. See UBI et al. Role of SIRT1 in homologous recombination (2010) ” — Recent reports revealed that SIRT1 also deacetylates several DNA double-strand break (DSB) repair proteins. — Using nuclear foci analysis and fluorescence-based, chromosomal DSBrepair reporter, we find that SIRT1 activity promotes homologous recombination (HR) in human cells. Importantly, this effect is unrelated to functions of poly(ADP-ribose) polymerase 1 (PARP1), another NAD(+)-catabolic protein, and does not correlate with cell cycle changes or apoptosis.” That is, the effect is independent of the PARP-related mechanism for repairing single-stranded breaks.
Major SIRT1 functions: #6 – deacetylation nonhistone proteins – transcription factors, co-activators, co-repressors, methyl binding proteins, etc.
In addition to the effects of SIRT1 on histones, p53, and DNA repair, SIRT1 deacetylates many other proteins within the nucleus. Most of these are transcription factors or co-activators which increase gene expression. They include the ones listed in this diagram:
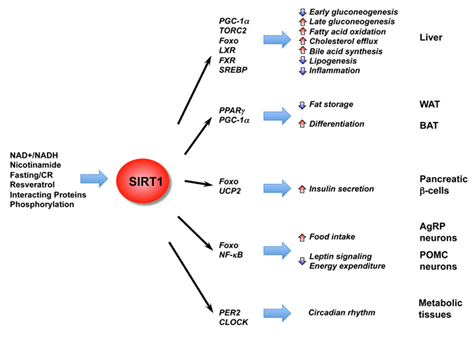
Image and legend source: Mammalian Sirtuins and Energy Metabolism “ The diverse functions of SIRT1 in central nutrient sensing and peripheral energy metabolism. The activity of SIRT1 is regulated by the cellular metabolic status, small molecule activators, interacting proteins, as well as post-translational modifications. After activation, SIRT1 modulates a variety of metabolic activities systemically and locally through either direct protein deacetylation or indirect chromatin remodeling.”
Following is a list of the non-histone protein targets of SIRT1 shown above, along with several other CR proteins not shown in the diagram. SIRT1 deacetylates over a dozen non-histone proteins, including p53, HSF1, eNOS, STAT3, FOXOs, PGC-1a, PPARγ, LXR, NF-kB, PER2, CLOCK, UCP2, MyoD. Other effects of SIRT1 are paradoxic are the effects of SIRT1 on FOXA2, LKB1, and Nrf2 (E2F1).
p53Ac – The most important non-histone target of SIRT1 is the protein called p53 already mentioned here. Both SIRT1 and SIRT2 deacetylate p53 and thereby increase cell survival. SIRT1 deacetylates lysine 382 (K382) on the SIRT1 protein. On the other hand, inhibiting both SIRT1 and SIRT2 (but not one of these two SIRTs) induces cell death in both cancer cells and non-cancer cells. Thus p53 has often been referred to as the “master tumor suppressor” or the “guardian of the genome”. Regardless of the terminology describing p53, it is clear that both SIRT1 and SIRT2 deacetylate p53 and promote cellular survival. This is one of the most important functions of SIRT1/2 and is a fundamental reason why Sirtuins probably promote longevity (Peck, SIRT Inhibitors Induce Cell Death and p53 Acetylation through Targeting Both SIRT1 and SIRT2 2010).
HSF1Ac – Heat shock factor 1 is a thermally sensitive protein that trimerizes migrates into the nucleus in response to heat stress or oxidative stress.
LXRAc – Caloric restriction has a positive effect on lowering cholesterol. This is due to the fact that SIRT1 deacetylates the nuclear receptor liver X receptor (LXR) protein
FXRAc - The farnesoid X receptor regulates the expression of a number of its target genes(ref).
eNOSAc – Reduced caloric intake decreases arterial blood pressures in healthy individuals and improves endothelium-dependent vasodilation in obese individuals. Endothelial nitric oxide synthetase (eNOS) is an enzyme that makes nitric oxide from L-arginine, relaxes blood vessels and thereby lowers blood pressure with CR. L-arginine is a popular “anti-aging” supplement that is marketed as a “CR mimetic”, but has failed to lengthen life span or increase health span. The likely reason for the “arginine failure” is that SIRT1 is needed to remove the “acetyl group” from lysines 496 (K496) and 506 (K506) from eNOS. This can only be accomplished by activating SIRT1 with CR or fasting. However, with old age, there is inadequate NAD+ to drive SIRT1 deacetylation. It is for this reason that we predict that NAD+ or NAD+ precursors like NMN will lower blood pressure in healthy individuals and improve flow-mediated dilation (FMD) in both healthy and obese individuals.
FMD (flow-mediated dilation) can easily be measured with clinically validated testing devices such as the EndoPAT or the VENDYS systems, which use a brachial blood pressure cuff and finger sensors which measure reactive hyperemia index (EndoPAT) or finger temperature warming (VENDYS). Both systems are clinically validated as non-invasive measurements of eNOS activity.
FOXAAc – The FOXA symbol in the diagram above is an upstream event that activates Glucagon signaling and inhibits Insulin signaling. Reduced caloric intake reduces insulin levels yet increases insulin sensitivity, increases glucagon production, and improves pancreatic β-cell function via the forkhead transcription factor called FOXA2. FOXA transcription factors play a key role in gene regulation of genes involved with glucose metabolism by binding to promoter regions on these genes with SIRT1. When nutrients are scare or when cells are starved, FOXOA2 can be acetylated in at least 7 different lysine residues. This increases FOXOA2 protein stability. SIRT1deacetylates these lysine residues in times of CR or starvation. NAD+ depletion disrupts this SIRT1-mediated stress response and results in excessive accumulation of FOXOA2 proteins. Restoring nuclear NAD+ levels should restore the normal interaction between FOXOA2 and SIRT1. This could be measured with oral glucose tolerance testing (OGTT), insulin tolerance testing (ITT), fasting blood glucose and fasting insulin, as well as HbA1c and Glycomark testing.
FOXOAc – Caloric restriction, fasting, and reducing Insulin/IGF-1 pathway signaling all promote the nuclear localization of the Forkhead homeobox type O transcription factors (FOXOs). FOXOs play a major role in cellular stress resistance by turning on genes that code for anti-oxidant enzymes, such as MnSOD (SOD2) and catalase. FOXOs also control genes for apoptosis. Unfortunately, none of these benefits occur unless blood sugar levels are low and insulin/IGF-1 signaling is low. In these conditions, the FOXOs can all migrate into the cell nucleus and turn on many genes responsible for cellular stress resistance. Both SIRT1 and SIRT2 deacetylate FOXO transcription factors and thereby activate the FOXOs. For instance, SIRT1 deacetylates FOXO1 which then increases gluconeogenesis in the liver. SIRT2 deacetylates FOXO3a and also promotes hepatic gluconeogenesis.
We predict that restoring nuclear NAD+ levels with NAD+ precursor therapy will increase cellular stress resistance and this could be measured by stem cell survival with stem cell harvesting, stem cell transplantation, and stem cell banking (protection from freeze/thawing induced apoptosis). This could be easily measured in the laboratory by culturing stem cells under conditions of cellular stress.
LKB1Ac – Liver kinase B1 (LKB1) is an enzyme that mediates much of the effect of mitochondrial biogenesis with CR or fasting. This occurs due to the fact that LKB1 is a direct activator of AMPK, the master regulator of metabolism and mitochondrial biogenesis. Recently, it has been disclosed that LKB1 also functions as a tumor suppressor by maintaining epithelial integrity. In tumors where LKB1 is mutated, cells loose an orderly epithelial configuration and the cancers start to grow rapidly LKB1 must be deacetylated by SIRT1 for proper tumor suppression and for AMPK activation. SIRT1 deacetylates lysine 48 (K48) on LKB1 and causes the LKB1 to leave the nucleus and go to the cytoplasm where it can associated with the adaptor protein STE20, activating itself and AMPK.
Thus, SIRT1 deacetylation of LKB1 induces LKB1 and AMPK activity in the cytoplasm. (Ruderman, SIRT1 Modulation of the Acetylation Status, Cytosolic Localization, and Activity of LKB1 2008). Both of these effects promote longevity and increase energy. For this reason, we predict that restoring nuclear NAD+ levels will have a direct effect on reducing age-induced fatigue. This could be measured with a VO2 max measuring device and endurance testing. We also predict that restoring nuclear NAD+ levels will re-organize skin cells that have lost their epithelial polarity, restoring better looking skin and improving skin histologic architecture on light microscopy.
MyoDAc – MyoD is a key transcription factor in skeletal muscle differentiation.
STAT3Ac – Skeletal muscle insulin resistance is a key component of the underlying cause of type II diabetes. Fasting and CR both can increase whole body insulin sensitivity, even when practice for brief periods of time (4-20 days). Although there are multiple ways that CR changes Insulin/IGF-1 signaling, the primary method by which CR promotes insulin sensitivity appears to be via the (inhibition of ) STAT3. SIRT1 deacetylates STAT3, which inactivates the transcription factor. This reduces gene expression for two subunits of the enzyme PI3K (p55a/p50a). This results in more efficient PI3K signaling with insulin stimulation. In summary, SIRT1 inhibits STAT3 which inhibits gene expression for PI3K subunits, thereby increasing energy expenditure.
IRS1/IRS2Ac – CR has a paradoxic effect on insulin receptor substrate 1 and 2 (IRS1/IRS2) gene expression. IRS1 and IRS2 are the first proteins in the molecular cascade of events that occurs when Insulin binds to the Insulin/IGF receptor on the cell. With CR, however, there is an increase in protein expression of IRS1/IRS2 in muscle, which in turn increases intracellular signaling for the Insulin/IGF-1 pathway. SIRT1 deacetylates IRS1/IRS2, thereby “turning off” this pro-aging pathway.
Nrf2Ac (E2F1) – Sirtuins have a paradoxic effect on the transcription factor called Nuclear factor Erythroid 2-related factor 2 (Nrf2 or E2F1). Normally, cellular stress activates the nuclear localization of Nrf2 which then in turn activates gene transcription by binding to the antioxidant response element (ARE) found in the promoter sites of these genes. This can occur due to direct effect of ROS on Nrf2 binding to its binding partner, Keap-1. It can also occur by the acetylation of Nrf2 by p300/CBP (aka CREB-binding protein). Paradoxically, both SIRT1 and SIRT2 can deacetylate Nrf2, SIRT1 deacetylates at two specific lysine residues (K588 and K591) which promotes cytoplasmic localization of the Nrf2 protein and prevents nuclear localization of Nrf2. As a result, SIRT1 actually is an inhibitor of Nrf2 antioxidant gene transcription. This is one of the paradoxic effects of SIRT1 and CR on Nrf2 (Kawai, Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization 2010).
Another paradoxic effect is the effect of SIRT2 on Nrf2. SIRT2 also deacetylates Nrf2 and therefore can decrease Nrf2 gene expression. One of these genes is the major cellular iron exporters called FPN1. Nrf2 activates FPN1 gene expression and SIRT2 inhibits FPN1 gene expression (Yang, Sirtuin 2 mediated-deacetylation regulates cellular iron homeostasis
2014). Thus SIRT2 controls regulates iron levels within the cell, decreasing iron export by FPN1, by deacetylating Nrf2.
p66shc – The adaptor protein p66shc is a major “pro-aging” factor within the cell. When growth factor signaling or ROS activates p66shc, the protein is phosphorylated on Serine (S). This promotes mitochondrial migration of p66shc into the mitochondrial matrix where it increases ROS production from mitochondria. SIRT1 decreases p66shc activity by decreasing both p66shc mRNA and p66shc protein levels. The molecular mechanism responsible for SIRT1 repression of p66shc gene expression is thought to be mediated by the epigenetic silencing of the p66shc gene by SIRT1-mediated histone deaetylation. Thus SIRT1 is a negative regulator of ROS production by epigenetically repressing p66shc gene expression (Xu, Salvianolic acid A preconditioning confers protection against concanavalin A-induced liver injury through SIRT1-mediated repression of p66shc in mice 2013) (Paneni, Molecular pathways of arterial aging 2015).
PGC-1αAc – Peroxisome proliferator activated receptor PPAR-γ co-activator-1α (PGC-1 α) is a master gene co-activator that works with over a dozen different transcription factors to turn on hundreds of genes involving peroxisome and mitochondrial biogenesis. Thus, PGC-1α turns on all of the genes involving making and burning of fat, producing of ketones, making ATP, and generating energy. SIRT1 also promotes gluconeogenesis in the liver by deacetylating PGC-1α (along with deacetylating FOXO1). Earlier blog entries discussing PGC-1 α can be found in this list,
PPARγ – Caloric restriction also activates the master transcription factor for the production of peroxisomes, called peroxisome proliferator activator receptor gamma (PPARγ). PPARγ induces long chain fatty acid oxidation, which is the first step required before fats can be oxidized in the mitochondria by beta-fatty acid oxidation. SIRT1 deacetylates PPARγ and then recruits a coactivator for PPARγ called Prdm16. Then the PPARγ/Prdm16 complex can activate the genes required to make brown fat. You can see the blog entry Getting skinny from brown fat,
NF-kβ – Nuclear factor kappa-β is a transcription factor that is the “master switch” for inflammation. SIRT1 deacetylates NF- kβ at the p65 subunit of NF-kB at lysine 310. This “turns off” gene expression for all of the genes involving inflammation, such as CRP, TNF- α, IL-1β, etc. Since arthritis is a universal feature of aging and all of these inflammatory biomarkers are elevated in joints with OA, we predict that restoring nuclear NAD+ with NAD+ precursor therapy will reduce the signs and symptoms of OA. This could be measured with various validated clinical scoring systems, such as the K-L scoring system based on plain, X-rays with weight bearing, the WOMAC scale, the IKDC scale, etc. All of these should improve with NMN or other NAD+ enhancing therapy.
Fine Tuning of SIRT1 Protein Activity
Endogeneous Activators and Inhibitors of Sirtuins – NAD+, NAM, AROS, lamin A, Tenovins, DBC1/CCAR2, CD38, HIC1, Cathepsin D
Alas, in biology for any generalization there is too often one or more “An the other hands, —.” We have discussed SIRT1expression here as if it were mainly driven by NAD+ level. Actually, it is affected both positively and negatively by many interacting variables. SIRT1 activity is increased by increasing nuclear NAD+, by fasting, and by caloric restriction. Most of these positive effects on SIRT1 are mediated by increases in nuclear NAD+ levels. SIRT1 activity is also inhibited by nicotinamide. This may be why niacin supplementation has not been shown to have a longevity effect, since it is converted into nicotinamide within the cell. In addition to activating SIRT1 with NAD+, there are naturally derived exogenous compounds such as reseveratrol and other plant-based compounds that bind to a different site on SIRT1, increasing it’s activity independently from NAD+. There are also several exogenous small molecule inhibitors of SIRT1, such as Tenovin-1 and Tenovin-6 which prevent the deacetylase activity of SIRT1. There are also endogenous proteins that bind directly to SIRT1 and increase or decrease the activity of SIRT1, such as AROS, lamin A, and DBC1. AROS is a protein that binds to SIRT1 and increases its activity. DBC1 (aka CCAR2) is a protein that binds to SIRT1 and inhibits SIRT1. We discussed these two in the Part 1 blog entry. HIC1 is a protein that binds to the SIRT1 promoter and increase SIRT1 gene expression. Another recently discovered protein that binds to SIRT1 is lamin A, a nuclear cytoskeletal protein that binds to SIRT1 and activates the protein. Another endogenous inhibitor of SIRT1 is the enzyme CD38. Although initial reports labeled CD38 as an “ectoenzyme”, several reports have shown that the CD38 protein is found on the inner membrane of the nucleus of the cell. Aksoy and colleagues have shown that CD38 is an SIRT1 inhibitor (Aksoy, Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38 2006).
In inflammatory joint diseases such as osteoarthritis and rheumatoid arthritis, there is another endogenous disruptor of SIRT1 – Cathepsin B. Normally SIRT1 has a positive effect within the joint, inhibiting chondrocyte apoptosis and promoting extracellular matrix synthesis of proteins such as alpha2(I) collagen. However, when there are high levels of TNF-a within the joint, TNF-a mediates a Cathepsin-B induced cleavage of the SIRT protein at amino acid 533, creating a 75-kd SIRT1 fragment that is no longer functional. As a result, there are low levels of SIRT1 activity within the chondrocytes and synovial cells of arthritic joints.
Some of these activators and inhibitors of SIRT1 activity are illustrated below:
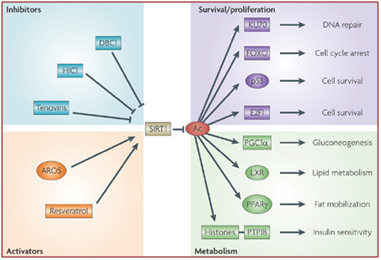
Image source: How does SIRT1 affect metabolism, senescence and cancer?
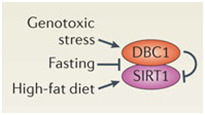
DBC1 (aka CCAR2) is one of the primary inhibitors of SIRT1 within the nucleus and it is activated by DNA damage (genotoxic stress). Fasting also inhibits the interaction of DBC1 and SIRT1. This may be a NAD+-independent molecular mechanism for how fasting increases SIRT1 activity. Likewise, a high fat diet activates SIRT1 binding to DBC1. This may be an NAD+-independent effect of a high-fat diet inhibiting Sirtuins.
SIRT1 Phosphorylation – SIRT1 phosphorylation can increase or decrease SIRT1 activity and SIRT1 half life
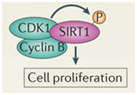
In addition to the above factors, SIRT1 is modified by at least 7 enzymes that phosphorylate SIRT1 at 13 “site-specific” locations on the SIRT1 protein. These enzymes phosphorylate different sites, and thereby either inactivate SIRT1 (AMPK), activate SIRT1 (cAMP/PKA, Cyclin B/Cdk1, CK2, JNK1, DYRKs) or increase the half life of the SIRT1 protein (JNK2). For instance AMPK phosphorylates SIRT1 at Thr344, which inactivates the p53 deacetylase function of SIRT1. On the other hand, DYRKs (Dual specificity tyrosine phosphorylation-regulated kinases) phosphorylate SIRT1 at Thr522 in response to environmental stress. This DYRK-mediated phosphorylation prevents SIRT1 from forming oligomeric aggregates of SIRT1 proteins. As a result, the Thr522 phosphorylated SIRT1 forms only monomers and thereby becomes a more active deacetylator of p53. Thus SIRT1 phosphorylation by AMPK and DYRKs have opposite effects on SIRT1 deacetylation of p53. Two phosphorylation sites on SIRT1 help regulate the cell cycle. Specifically, the CyclinB/Cdk1 complex phosphorylates SIRT1 at Thr530 and Ser540. Thus CyclinB/Cdk1 phosphorylation of SIRT1 increases cell proliferation. This is why SIRT1 is so important for cell growth and is present in high levels in mitotically active cells. Even the two cJun N-terminal kinases, JNK1 and JNK2, have different phosphorylation sites on SIRT1 and different effects. For instance, JNK1-mediated phosphorylation of the Ser47 residue on SIRT1 increases the histone 3 (H3) deacetylase activity of SIRT1, whereas JNK2-mediated phosphorylation of the Ser27 residue of SIRT1 increase the half life of the SIRT1 protein from 2 hours to 9 hours. Thus each of these different “SIRT1 phosphorylators” add a phosphate group at specific sites on SIRT1. This increase specific activities of SIRT1 and these effects are independent of NAD+ levels within the cell. Last of all, UV light and free radicals such as hydrogen peroxide (H2O2) inactivates SIRT1 by another protein called SENP. SENP inactivates SIRT1 by removing a sumoyl group from the SIRT1 protein. Here are some diagrams that illustrates how “SIRT1 phosphorylators” activate SIRT1 activity and how de-sumoylation of SIRT1 inhibits SIRT1.
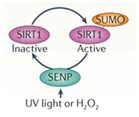
De-SUMOylating SIRT1
UV light or H2O2 inactivate SIRT1 via a UV/ROS sensitive protein called SENP. SENP removes a SUMO group from SIRT1, thereby inactivating the protein.
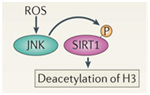
ROS activation of SIRT1
Free radicals such as H2O2 or superoxide can activate the SIRT1 protein via a protein kinase called c-Jun Kinase (JNK), increasing the ability of SIRT1 to deacetylate histone proteins such as H3.
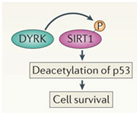
SIRT1 and Cell Survival
DYRKs are enzymes that are activated by ROS or (environmental stress) or DNA damage. DYRKs activate SIRT1-mediated deacetylation of p53, thus increasing cell survival in stressful times.
SIRT1 and the Cell Cycle
Two proteins involved with cell cycle regulation form a complex with SIRT1 and phosphorylate the SIRT1 protein. This increases cell proliferation and is a key factor in allowing mitotic cells to keep dividing.
In summary, SIRT1 activity is dependent on NAD+ levels, nicotinamide levels, phosphorylation status, DBC-1 status, AROS status, ROS or other stressors, and the presence of exogenous SIRT1 activators (resveratrol) or inhibitors (Tenovins).
The over-all importance of Sirtuins, however, is their ability to affect gene expression in the nucleus. Changing gene expression is is considered an “upstream event” in aging. In this aspect, SIRT1 is not the only Sirtuin in the nucleus of the cell. There are two others that we will discuss now – SIRT6 and SIRT7.
SIRT6 – The Master DNA Repair and Genomic Stability Sirtuin
The effects of SIRT6 on Genome Stability
Another Sirtuin family member that directly affects gene expression is SIRT6. SIRT6 is also found in the nucleus and is also an NAD+-dependent enzyme. Like SIRT1, SIRT6 plays a vital role in both single strand DNA breaks (SSBs) and double stranded DNA breaks (DSBs). With DSBs, SIRT6 is one of the earliest factors that is recruited to the site of the DSB. This occurs within 5 seconds in a healthy cell. SIRT6 recruits the chromatin remodeler, SNF2H, to the site of the DSB and deacetylates histone 3 at lysine 56 (H3K56). This allows SNF2H to open up the chromatin at the DSB site, which then allows the DNA DSB repair proteins to be recruited (BRCA1, 53BP1, and RPA). This is shown in the diagram below.
SIRT6 – The Early Responder to DSBs
Double stranded DNA breaks are the most lethal DNA damage type and are primarily responsible for cancer and aging. Of the seven SIRTs, the SIRT6 isoform probably plays the greatest role in DNA repair. In the diagram that follows, a double stranded DNA break occurs and within 5 sec, SIRT6 shows up to deacetylate H3K56. This recruits the chromatin remodeler, SNF2H, which then “opens up” the chromatin, allowing the key DNA repair proteins to bind to the site of injury. The key DNA repair proteins include 53BP1, RPA, BRCA1, and CtIP (not shown). Collectively, these proteins repair double-strand DNA breaks by the more accurate repair program called “homologous recombination”, or HR. The other DNA DSB break repair pathway, NHEJ, is not as accurate a repair pathway as the HR pathway, which is the pathway shown in the diagram.
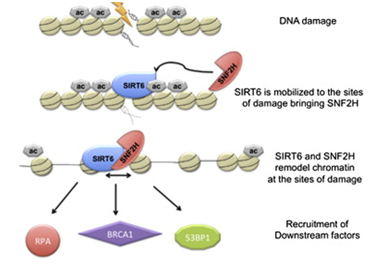
Image and highlights source SIRT6 Recruits SNF2H to DNA Break Sites, Preventing Genomic Instability through Chromatin Remodeling “Highlights: •SIRT6 arrives to DNA break sites within 5 s, •Lack of SIRT6 and SNF2H increases sensitivity to genotoxic damage, •SIRT6 directly recruits SNF2H, which in turn opens chromatin at break sites. •SIRT6 and SNF2H are necessary to recruit 53BP1, RPA, and BRCA1 to the damage sites”
With DSBs, SIRT6 plays crucial roles in both the nonhomologous end joining (NHEJ) pathway and the homologous recombination (HR) pathway. With HR, SIRT6 deacetylates a key protein called CtIP which works with the BRCA1 protein to generate single stranded DNA (aka “DNA end resection”) at the site of the DSB. Unless CtIP and BRCA1 can form a single strand of DNA at the break site by “DNA end resection”, then the non-homologous end joining (NHEJ) method of DNA repair takes over, which does not repair DSBs nearly as accurate as HR. In addition to the effects of SIRT6 on HR and NHER repair of DSBs, SIRT6 activates PARP1 directly as well. This increases DNA repair for single stranded DNA breaks (SSBs) by increasing base-excision repair (BER) pathway for SSBs and also increases double stranded DNA repair by both HR and NHEJ pathways. All of these effects have a net effect of increasing genome stability within the cell.
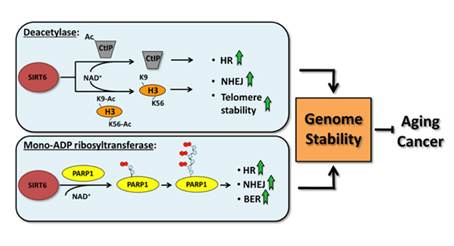
Image and legend source: 2011 Repairing split ends: SIRT6, mono-ADP ribosylation and DNA repair “SIRT6 regulates genomic stability. SIRT6 promotes genome stability by regulating DNA single-strand and double strand break repair pathways and by facilitating telomere maintenance. The deacetylase and the mono-ADP ribosyltransferase activities of SIRT6 both contribute to this function.”
Based on the three mechanisms illustrated above, SIRT6 plays a key role in both SSB and DSB repair. As a result, when SIRT6 knock-out mice are created, they have an accelerated aging-like phenotype.
The Other Effects of SIRT6 besides Genome Stability
Other than genomic stability, the other primary effect of SIRT6 are mediated by histone protein modifications of specific lysines found on Histone 3 (H3K9 and H3K56). Although SIRT1 has some histone protein deacetylase function, it has a much greater mono-ADP-ribosyltransferase activity and an even greater “deacylation” function. “Deacylation” refers to the removal of long chain fatty acids (palmitic acid, myristic acid, oleic acid, and linoleic acid) from lysine amino groups on proteins. One of the SIRT6 targets for ADP-ribosylation is PARP1 (see above). PARP1 is mono-ADP-ribosylated by SIRT6 which activates PARP1. The “deacylation” function of SIRT6 was just recently discovered. Here is a diagram that shows the effects of SIRT6 deacetylase function and the effects of SIRT6 on CtIP which also occurs via its effects on gene expression in the nucleus of every cell. (The mono-ADP-ribosyltransferase and deacylation function of SIRT6 are not shown in the diagram).
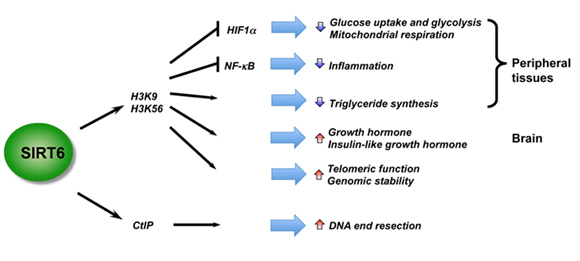
Illustration and legend reference “SIRT6 is a critical regulator in genome stability, metabolism, and inflammatory response. By deacetylation of H3, SIRT6 regulates metabolic homeostasis and inflammatory response in peripheral tissues, while functioning as a central regulator of somatic growth.”
As you can see, most of the effects of SIRT6 are mediated by histone protein deacetylation of two specific lysine residues found on the histone 3 (H3) subunit of the histone “spools” that wind up DNA and compact it into the nucleus. The H3 subunit has two specific lysine residues found on the tail of “H3” protein called H3K9 and H3K56. When these two sites are deacetylated by SIRT6, this inhibits the type of metabolism seen in cancer cells called “Warburg metabolism” where the cells are totally dependent on sugar for generating energy and cannot burn fats. Also, SIRT6 inhibits the inflammation caused by the major inflammatory “on switch” called NF-kB. The other mechanisms of action of SIRT6 have not been fully elucidated, such as the effects of SIRT6 on growth hormone, IGF-1 and telomere stability. What appears clear, however, is that all of the effects of SIRT1 and SIRT6 occur in the nucleus of the cell and all of the activity of both SIRT1 and SIRT6 are dependent on the availability of NAD+ in the nucleus of the cell.
The effects of SIRT7 within the cell, SIRT7 activators and SIRT7 inhibitors
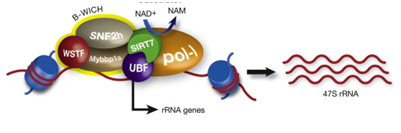
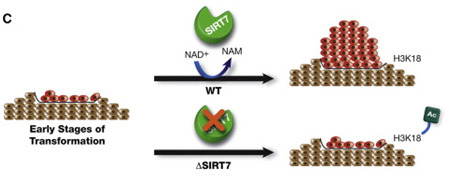
Image source: A Big Step for SIRT7, One Giant Leap for Sirtuins… in Cancer
Outlier Research
Unfortunately, there are research results that challenge some of the basic assumptions of those who are looking for health creation through enhancing body NAD+ levels. And, they chellenge some of our predictions listed above. For example there seems widespread agreement that:
- If you want to activate SIRT1, you must have adequate levels of nuclear NAD+.
2, Having adequate levels of nuclear NAD+ is the best way to keep SIRT1 active.
- The impact of SIRT1 is universally health-producing.
Unfortunately, there appears to be research that suggest that these broad assertions ain’t always necessarily so. We have above discussed how SIRT1 phosphorylation can increase or decrease SIRT1 activity and a number of proteins that bind to SIRT1 and increase or decrease its activity. Cold is another example.
Cold can activate SIRT1 in a way that is completely independent of NAD+
You can use cold to activate SIRT1 and PGC1alpha regardless of your nuclear NAD+ status. The December 2011 article The cAMP/PKA Pathway Rapidly Activates SIRT1 to Promote Fatty Acid Oxidation Independently of Changes in NAD+ reports: “Highlights
- Stimulation of the cAMP/PKA pathway results in phosphorylation of SIRT1 serine 434
- SIRT1 S434 phosphorylation increases intrinsic deacetylase activity
- SIRT1 activation by S434 phosphorylation is rapid and independent of changes in NAD+
- S434 phosphorylation induces PGC-1α deacetylation and increased fatty acid oxidation”
“The NAD+-dependent deacetylase SIRT1 is an evolutionarily conserved metabolic sensor of the Sirtuin family that mediates homeostatic responses to certain physiological stresses such as nutrient restriction. Previous reports have implicated fluctuations in intracellular NAD+concentrations as the principal regulator of SIRT1 activity. However, here we have identified a cAMP-induced phosphorylation of a highly conserved serine (S434) located in the SIRT1 catalytic domain that rapidly enhanced intrinsic deacetylase activity independently of changes in NAD+ levels. Attenuation of SIRT1 expression or the use of a nonphosphorylatable SIRT1 mutant prevented cAMP-mediated stimulation of fatty acid oxidation and gene expression linked to this pathway. Overexpression of SIRT1 in mice significantly potentiated the increases in fatty acid oxidation and energy expenditure caused by either pharmacological β-adrenergic agonism or cold exposure. These studies support a mechanism of Sirtuin enzymatic control through the cAMP/PKA pathway with important implications for stress responses and maintenance of energy homeostasis.”
We have discussed cold as a simple and practical hormetic healh intervention in previous blog entries(ref)(ref)(ref)(ref).
SIRT1 turns off Nrf2 and its health-producing affects
We mentioned this above. We have published several blog entries on the numerous impacts of activating Nrf2 which turns on hundreds of positive “antioxidant response genes.” See this list. One of the last things we would expect to find is that SIRT1 which also produces numerous health benefits actually turns Nrf2 off. but that appears to be the case. It tends to keep Nrf2 in the cytoplasm rather than allowing it to migrate to the nucleus where it can do good. Even worse, resveratrol, our favorite SIRT1 activator actually inhibits expression of Nrf2.
SIRT1 “turns off” Nrf2-mediated antioxidant gene expression?
The 2010 publication: Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization reports: “Activation of Nrf2 by covalent modifications that release it from its inhibitor protein Keap1 has been extensively documented. In contrast, covalent modifications that may regulate its action after its release from Keap1 have received little attention. Here we show that CREB-binding protein induced acetylation of Nrf2, increased binding of Nrf2 to its cognate response element in a target gene promoter, and increased Nrf2-dependent transcription from target gene promoters. Heterologous sirtuin 1 (SIRT1) decreased acetylation of Nrf2 as well as Nrf2-dependent gene transcription, and its effects were overridden by dominant negative SIRT1 (SIRT1-H355A). The SIRT1-selective inhibitors EX-527 and nicotinamide stimulated Nrf2-dependent gene transcription, whereas resveratrol, a putative activator of SIRT1, was inhibitory, mimicking the effect of SIRT1. Mutating lysine to alanine or to arginine at Lys588 and Lys591 of Nrf2 resulted in decreased Nrf2-dependent gene transcription and abrogated the transcription-activating effect of CREB-binding protein. Furthermore, SIRT1 had no effect on transcription induced by these mutants, indicating that these sites are acetylation sites. Microscope imaging of GFP-Nrf2 in HepG2 cells as well as immunoblotting for Nrf2 showed that acetylation conditions resulted in increased nuclear localization of Nrf2, whereas deacetylation conditions enhanced its cytoplasmic rather than its nuclear localization. We posit that Nrf2 in the nucleus undergoes acetylation, resulting in binding, with basic-region leucine zipper protein(s), to the antioxidant response element and consequently in gene transcription, whereas deacetylation disengages it from the antioxidant response element, thereby resulting in transcriptional termination and subsequently in its nuclear export.”
A brand new 2015-dated publication seems to say the direct opposite to the just-cited publication Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating Sirt1 to resist AGEs-induced upregulation of fibronetin and transforming growth factor-β1 in rat glomerular messangial cells. “Highlights:
- PD increased Sirt1 levels, promoted Nrf2-ARE pathway activation, and reduced ROS levels in AGEs-treated GMCs. (PD is a resveratrol glycoside)
- PD resisted AGEs-induced upregulation of FN and TGF-β1 by activating Sirt1-Nrf2-ARE pathway.
- PD ameliorated DN in a STZ-induced diabetic rat model.”
“Sirt1 and nuclear factor-E2 related factor 2 (Nrf2)-anti-oxidant response element (ARE) anti-oxidative pathway play important regulatory roles in the pathological progression of diabetic nephropathy (DN) induced by advanced glycation-end products (AGEs). Polydatin (PD), a glucoside of resveratrol, has been shown to possess strong anti-oxidative bioactivity. Our previous study demonstrated that PD markedly resists the progression of diabetic renal fibrosis and thus, inhibits the development of DN. Whereas, whether PD could resist DN through regulating Sirt1 and consequently promoting Nrf2-ARE pathway needs further investigation. Here, we found that concomitant with decreasing RAGE (the specific receptor for AGEs) expression, PD significantly reversed the downregulation of Sirt1 in terms of protein expression and deacetylase activity and attenuated FN and TGF-β1 expression in GMCs exposed to AGEs. Under AGEs-treatment condition, PD could decrease Keap1 expression and promote the nuclear content, ARE-binding ability, and transcriptional activity of Nrf2. In addition, PD increased the protein levels of heme oxygenase 1 (HO-1) and superoxide dismutase 1 (SOD1), two target genes of Nrf2. The activation of Nrf2-ARE pathway by PD eventually led to the quenching of ROS overproduction sharply boosted by AGEs. Depletion of Sirt1 blocked Nrf2-ARE pathway activation and reversed FN and TGF-β1 downregulation induced by PD in GMCs challenged with AGEs. Along with reducing HO-1 and SOD1 expression, silencing of Nrf2 increased FN and TGF-β1 levels. PD treatment elevated Sirt1 and Nrf2 levels in the kidney tissues of diabetic rats, then improved the anti-oxidative capacity and renal dysfunction of diabetic models, and finally reversed the upregulation of FN and TGF-β1. Taken together, the resistance of PD on upregulated FN and TGF-β1 induced by AGEs via oxidative stress in GMCs is closely associated with its activation of Sirt1-Nrf2-ARE pathway.”
How do these and possibly many other contrarian or contradictory findings play out in-vivo? We simply don’t know. We don’t know how all the regulatory feedback inhibition loops will affect each other when we make a major intervention like enhancing body levels of NAD+. And we don’t know what the effect of many other variables will be, such as initial oxidative state, circadian state, relationship to stressors, disease states, hormonal states, age, sex, etc. Our theories which look at one pathway at a time under standardized conditions cannot tell us that. We will only know the effect of NAD+ enhancement therapy through observing net health effects and key health surrogate biomarkers. And we have to do that on and highly individualized basis That is why we regard the issue of individual biomarkers to have such great importance.
View the full article at Anti-Aging Firewalls












{kind=link}
{kind=link}
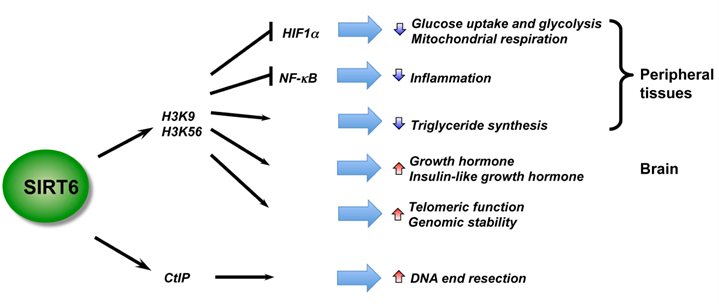{kind=link}